Tuberous sclerosis complex
Reading File
Finding Sources
Finding Sources
Searching Images
tuberous sclerosis complex features
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Tuberous Sclerosis Complex (TSC)
Overview
Tuberous sclerosis complex (TSC) is a multisystem disorder of cellular differentiation and proliferation, first described by Bourneville in 1880. It is characterized by the widespread development of hamartomas and benign neoplasms in the brain, skin, kidneys, heart, lungs, and eyes. Prevalence is approximately 1 in 5,000–6,000 individuals, though this is likely an underestimate given the variable expressivity. The classic triad of facial angiofibromas, epilepsy, and intellectual disability is present in only a minority of patients.
— Bradley and Daroff's Neurology in Clinical Practice; Robbins & Kumar Basic Pathology
Genetics and Molecular Pathogenesis
TSC is autosomal dominant with highly variable penetrance. The spontaneous mutation rate is remarkably high — 66–86% of cases arise de novo.
Two causative genes:
| Gene | Chromosome | Protein |
|---|---|---|
| TSC1 | 9q34.3 | Hamartin |
| TSC2 | 16p13.3 | Tuberin |
The hamartin–tuberin protein complex is a critical negative regulator of mTOR (mammalian target of rapamycin). It acts by downregulating the small G-protein RHEB (Ras homolog enriched in brain). Loss-of-function mutations in either gene cause constitutive mTOR overactivation → uncontrolled cell growth and hamartoma formation.
TSC2 vs. TSC1: TSC2 mutations predominate, especially in sporadic cases. TSC2 is associated with more severe disease — higher rates of intellectual disability, subependymal nodules, renal angiomyolipomas, and retinal phakomas. About 10–25% of patients have no identifiable mutation on conventional testing (some have mosaicism).
Importantly, TSC2 is adjacent to PKD1 on chromosome 16. Contiguous deletions inactivating both genes produce a combined phenotype of TSC and autosomal dominant polycystic kidney disease (ADPKD).
— Bradley and Daroff's Neurology in Clinical Practice; Harrison's Principles of Internal Medicine; Robbins & Kumar Basic Pathology
Diagnostic Criteria (2012 International Consensus)
Genetic Criterion
A pathogenic mutation in TSC1 or TSC2 alone is sufficient for a definite diagnosis.
Clinical Criteria
Major Features (11 total):
- Hypomelanotic macules (≥3, at least 5 mm diameter)
- Angiofibromas (≥3) or fibrous cephalic plaque
- Ungual (periungual) fibromas
- Shagreen patch
- Multiple retinal hamartomas
- Cortical dysplasias (tubers and white matter radial migration lines)
- Subependymal nodules (SENs)
- Subependymal giant cell astrocytoma (SEGA)
- Cardiac rhabdomyoma
- Lymphangioleiomyomatosis (LAM)
- Angiomyolipomas (≥2)
Minor Features:
- "Confetti" skin lesions
- Dental enamel pits (≥3)
- Intraoral fibromas (≥2)
- Retinal achromic patch
- Multiple renal cysts
- Non-renal hamartomas
Definite diagnosis: 2 major features, OR 1 major + ≥2 minor features
Possible diagnosis: 1 major feature, OR ≥2 minor features
Note: LAM + angiomyolipomas together, without other features, do not meet criteria for a definite diagnosis.
— Bradley and Daroff's Neurology in Clinical Practice
Clinical Manifestations by System
Skin (virtually universal)
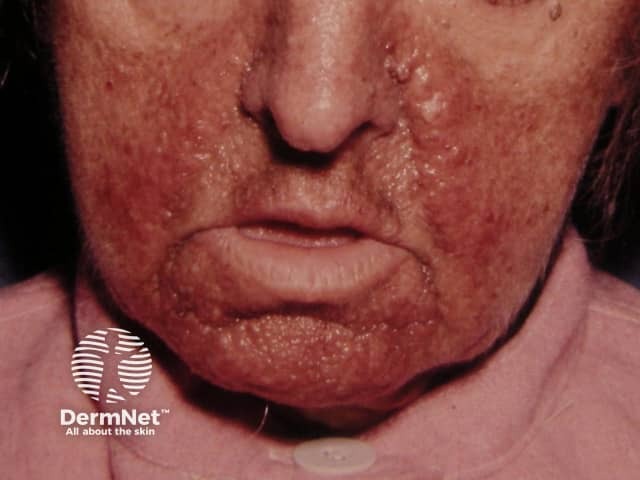
- Hypomelanotic macules (ash-leaf spots): Present in >90% of patients. Oval, leaf-shaped white macules, usually present at birth. May require Wood's lamp to visualize in fair-skinned infants. Also seen: linear macules and "confetti" stippled hypopigmentation.
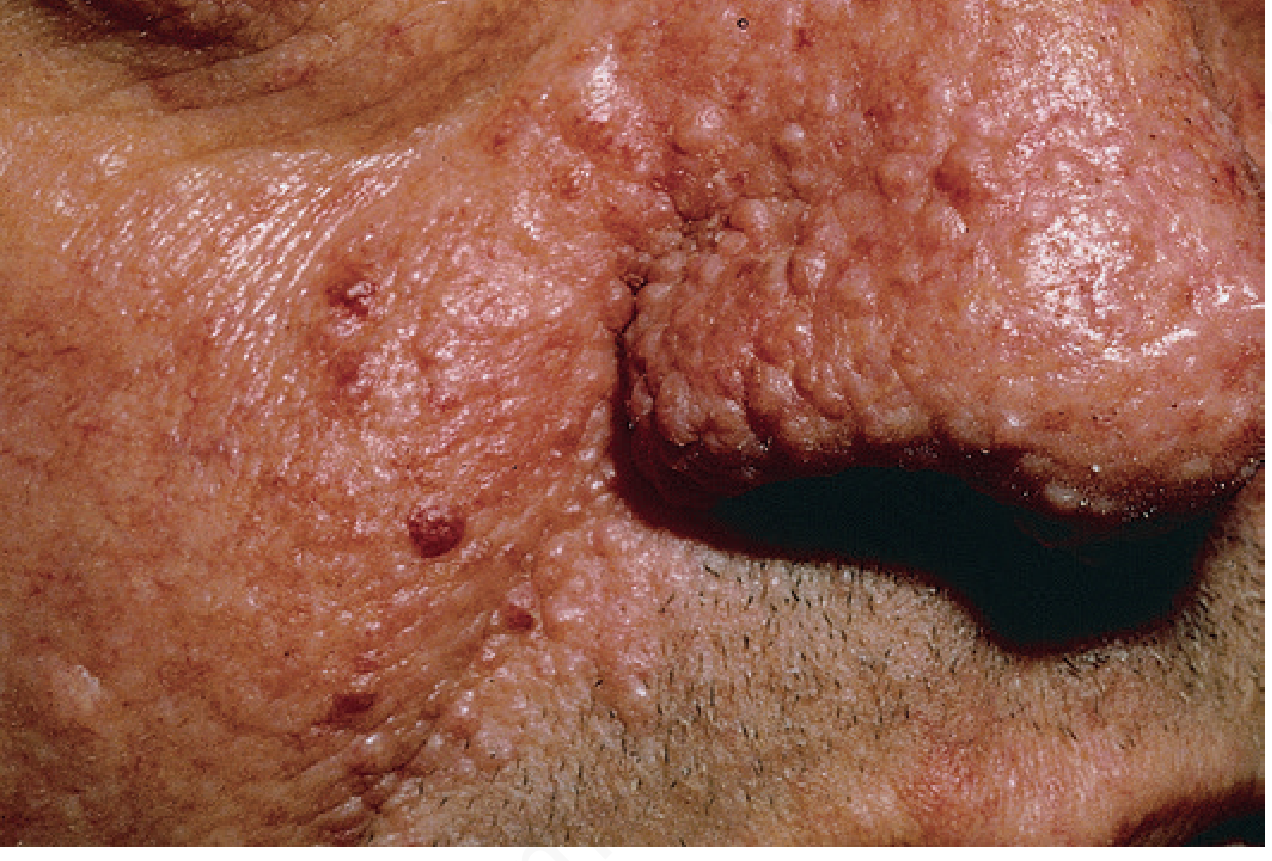
- Facial angiofibromas (adenoma sebaceum): Present in ~90% of patients >4 years old. Small (1–3 mm), yellowish-red, waxy papules distributed symmetrically over cheeks, nose, and forehead. Appear in preschool years and gradually increase in number.
- Shagreen patch: Leathery, cobblestoned, skin-colored plaque, typically in the lumbosacral region. Histologically a collagenoma.
- Ungual (periungual/subungual) fibromas: Flesh-colored growths erupting from nail folds.
- Fibrous cephalic plaque: Firm forehead or scalp plaque, histologically an angiofibroma.
- Café-au-lait spots and focal poliosis (white hair tufts) may also be present.
— Andrews' Diseases of the Skin; Bradley and Daroff's Neurology in Clinical Practice
Nervous System
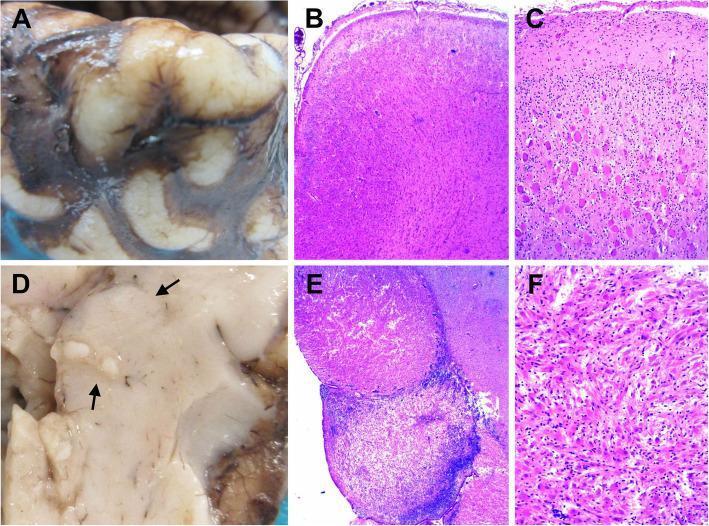
- Cortical tubers: Glioneuronal hamartomas causing focal epilepsy. Seizures are often refractory to antiepileptic drugs and are a major source of morbidity.
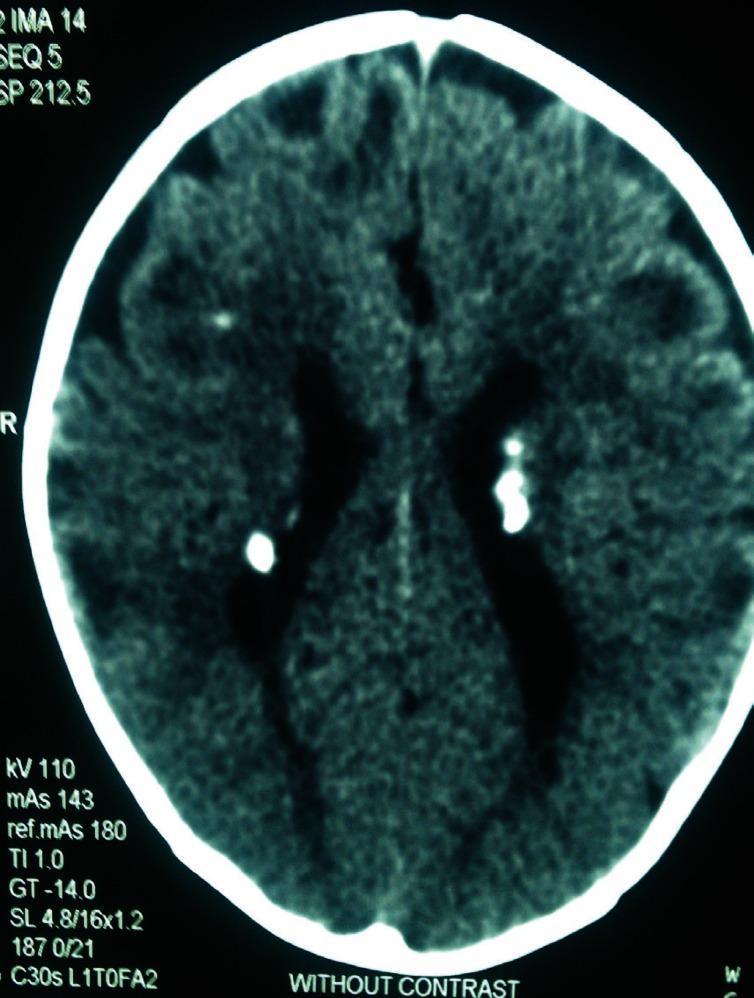
- Subependymal nodules (SENs): Calcified hamartomas lining the ventricles, often called "candle drippings" on imaging.
- Subependymal giant cell astrocytoma (SEGA): Benign glioneuronal tumor arising near the foramen of Monro. Causes obstructive hydrocephalus; may present acutely. Requires surgical intervention and/or mTOR inhibitor therapy.
- White matter radial migration lines: On MRI (FLAIR), linear hyperintensities extending from periventricular white matter to cortex — represent dysplastic cell migration tracks.
- Neuropsychiatric: Intellectual disability, autism spectrum disorder, attention deficit, anxiety. Collectively termed "TSC-associated neuropsychiatric disorders" (TAND). More common with TSC2 mutations.
— Robbins & Kumar Basic Pathology; Bradley and Daroff's Neurology in Clinical Practice
Kidneys
- Angiomyolipomas (AMLs): Most common renal finding. Multiple and bilateral. Usually benign but can hemorrhage spontaneously (Wunderlich syndrome). Prophylactic treatment recommended for lesions >4 cm.
- Renal cysts: Occur in 20–30% of patients. Radiographically similar to ADPKD cysts.
- Renal cell carcinoma: Clearly increased risk compared to the general population; requires regular periodic imaging surveillance.
- Chronic kidney disease: Rare but can progress to end-stage renal failure. Urine sediment typically unremarkable with only mild proteinuria.
— Harrison's Principles of Internal Medicine
Heart
- Cardiac rhabdomyomas: Most common cardiac tumor of childhood; often the presenting finding in infants (detected on fetal echocardiography). May cause arrhythmias or outflow obstruction. Most spontaneously regress.
Lungs
- Lymphangioleiomyomatosis (LAM): Proliferation of smooth muscle-like cells along lymphatics and airways. Predominantly affects women of reproductive age. Causes progressive dyspnea, chylothorax, and recurrent pneumothorax.
Eyes
- Retinal hamartomas (phakomas): Flat glial hamartomas or mulberry lesions at the optic disc or peripheral retina.
Pathology
The fundamental lesion is a hamartoma — a benign, disorganized overgrowth of tissue elements native to the site. Key histological hallmarks include:
- Balloon cells (giant cells with abundant glassy cytoplasm and eccentric nuclei) in cortical tubers and subependymal nodules
- Disorganized cortical architecture with loss of normal gray-white matter junction
The hamartin–tuberin complex normally functions as a GTPase-activating protein for RHEB, keeping mTOR inactive. Without this brake, mTOR drives unrestrained cellular anabolism, proliferation, and abnormal growth.
— Robbins & Kumar Basic Pathology; Bradley and Daroff's Neurology in Clinical Practice
Management
| Domain | Intervention |
|---|---|
| Seizures | Antiepileptic drugs (often difficult to control); vigabatrin is particularly effective for infantile spasms in TSC; surgery for refractory tuber-related epilepsy |
| SEGA | Surgical resection or everolimus (mTOR inhibitor); mTOR inhibitors shrink SEGA and may prevent hydrocephalus |
| Renal AMLs >4 cm | Selective embolization or nephron-sparing surgery; everolimus is FDA-approved for TSC-associated AMLs |
| LAM | Everolimus or sirolimus (mTOR inhibitors) — approved treatment; slows lung function decline |
| Facial angiofibromas | CO₂ or pulsed-dye laser; topical mTOR inhibitors (e.g., topical sirolimus) |
| Surveillance | Annual brain MRI (SENs/SEGA monitoring), renal imaging every 1–3 years, echocardiography, ophthalmology, neuropsychiatric assessment |
mTOR inhibitors (everolimus, sirolimus) are now considered first-line treatment for multiple TSC manifestations — renal AMLs, SEGA, LAM, and partially for epilepsy.
— Harrison's Principles of Internal Medicine; Bradley and Daroff's Neurology in Clinical Practice
Summary
TSC is a multisystem neurocutaneous syndrome driven by mTOR pathway overactivation from mutations in TSC1 or TSC2. Clinical expression is remarkably variable — from an incidental skin finding in an adult to devastating neonatal epilepsy. The skin nearly always provides the first diagnostic clue. Diagnosis relies on 2012 consensus criteria (genetic or clinical). The advent of mTOR inhibitors (everolimus, sirolimus) has transformed management, offering disease-modifying therapy across multiple organ systems simultaneously.
Tuberous sclerosis complex
Reading File
Finding Sources
Searching Images
tuberous sclerosis complex skin brain kidney features
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
I now have comprehensive content from all sources. Here is the full synthesis:
Tuberous Sclerosis Complex (TSC)
Overview
TSC is a multisystem disorder of cellular differentiation and proliferation, first described by Bourneville in 1880. It is characterized by hamartomas and benign neoplasms forming in the brain, skin, kidneys, heart, lungs, liver, and eyes. Prevalence is approximately 1 in 5,000–6,000, though likely an underestimate due to highly variable expressivity. The classic triad of facial angiofibromas, epilepsy, and intellectual disability occurs in only a minority of patients.
— Bradley and Daroff's Neurology in Clinical Practice; Andrews' Diseases of the Skin
Genetics and Molecular Pathogenesis
Inheritance: Autosomal dominant with variable penetrance. The spontaneous mutation rate is exceptionally high — 66–86% of cases arise de novo.
| Gene | Chromosome | Protein | Notes |
|---|---|---|---|
| TSC1 | 9q34.3 | Hamartin | Less severe phenotype |
| TSC2 | 16p13.3 | Tuberin | More severe; adjacent to PKD1 |
Mechanism: Hamartin and tuberin form a dimeric complex that negatively regulates mTOR by suppressing the small GTPase RHEB (Ras homolog enriched in brain). Loss of function in either → constitutive mTOR overactivation → uncontrolled cellular growth, abnormal proliferation, and hamartoma formation across tissues.
TSC2 vs TSC1:
- TSC2 mutations predominate, especially in sporadic cases
- TSC2 associated with more severe disease: greater rates of intellectual disability, subependymal nodules, renal angiomyolipomas, and retinal phakomas
- ~85% of clinically diagnosed patients have an identifiable mutation; the remainder may have mosaicism
- Contiguous deletions spanning TSC2 and the adjacent PKD1 gene produce a combined TSC + ADPKD phenotype
— Bradley and Daroff's Neurology in Clinical Practice; Harrison's Principles of Internal Medicine 22E; Robbins & Kumar Basic Pathology
Diagnostic Criteria (2012 International Consensus)
Genetic Criterion
A pathogenic mutation in TSC1 or TSC2 alone is sufficient for a definite diagnosis. Note: 10–25% of clinically confirmed TSC patients have no mutation on conventional testing — a negative result does not exclude TSC.
Clinical Criteria
Major Features (11):
- Hypomelanotic macules (≥3, diameter ≥5 mm)
- Angiofibromas (≥3) or fibrous cephalic plaque
- Ungual fibromas (≥2)
- Shagreen patch
- Multiple retinal hamartomas
- Cortical dysplasias (tubers and white matter radial migration lines)
- Subependymal nodules (SENs)
- Subependymal giant cell astrocytoma (SEGA)
- Cardiac rhabdomyoma
- Lymphangioleiomyomatosis (LAM)
- Angiomyolipomas (≥2)†
Minor Features:
- "Confetti" skin lesions
- Dental enamel pits (≥3)
- Intraoral fibromas (≥2)
- Retinal achromic patch
- Multiple renal cysts
- Non-renal hamartomas
Definite diagnosis: 2 major features, OR 1 major + ≥2 minor features
Possible diagnosis: 1 major feature, OR ≥2 minor features
†LAM + angiomyolipomas together, without other features, do not alone meet criteria for definite diagnosis.
— Bradley and Daroff's Neurology in Clinical Practice
Clinical Manifestations
Skin (virtually universal)
| Lesion | Description | Frequency |
|---|---|---|
| Hypomelanotic macules (ash-leaf spots) | Oval, leaf-shaped white macules; may require Wood's lamp in fair skin; also linear and "confetti" variants | >90% |
| Facial angiofibromas (adenoma sebaceum) | 1–3 mm, yellowish-red, waxy papules; bilateral, symmetric over cheeks, nose, forehead; appear in preschool years | ~90% of patients >4 yrs |
| Shagreen patch | Irregularly shaped, textured, flesh-colored plaque; typically lumbosacral; histologically a collagenoma | 20–30% |
| Ungual/periungual fibromas | Fleshy nodules adjacent to or beneath nails; present in up to 80% of older adults | ~20% overall |
| Fibrous cephalic plaque | Firm forehead/scalp plaque; histologically an angiofibroma | Variable |
| Focal poliosis | White hair tufts at birth | Variable |
— Andrews' Diseases of the Skin; Bradley and Daroff's Neurology in Clinical Practice
Nervous System
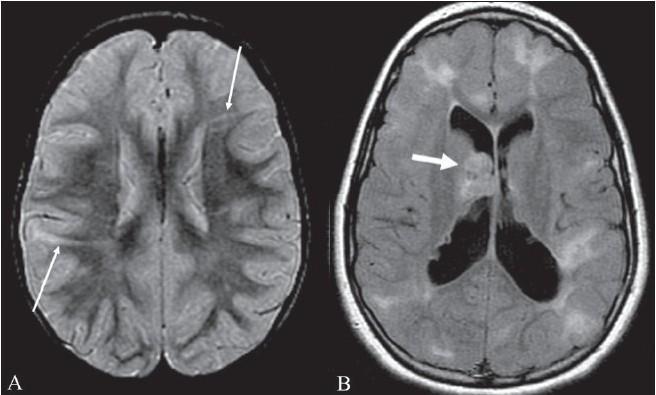
Cortical tubers: Focal glioneuronal hamartomas. Disrupt cortical lamination and gray-white matter junction; contain dysmorphic neurons and balloon/giant cells (large, eosinophilic, eccentric nuclei). Form between 14–16 weeks of gestation; tuber burden is set before birth. Tubers are the primary substrate for epilepsy.
Subependymal nodules (SENs): Calcified hamartomas lining the ventricular walls ("candle drippings" on imaging). Usually arise near the caudothalamic groove by the foramen of Monro. Calcify by adolescence and remain asymptomatic unless they transform into SEGA.
Subependymal giant cell astrocytoma (SEGA): Benign glioneuronal tumor arising near the foramen of Monro. Its location causes obstructive hydrocephalus, often presenting acutely. Requires surgical resection and/or mTOR inhibitor therapy.
White matter radial migration lines: Linear or wedge-shaped bands from ventricular surface to cortex, reflecting abnormal glial migration; visible on FLAIR/PD MRI.
Neuropsychiatric manifestations (TAND — TSC-Associated Neuropsychiatric Disorders):
- Intellectual disability (more common with TSC2)
- Epilepsy — frequently refractory; infantile spasms are common in infancy
- Autism spectrum disorder, ADHD, anxiety, mood disorders
- Behavioral disturbances
— Robbins & Kumar Basic Pathology; Bradley and Daroff's Neurology in Clinical Practice
Kidneys
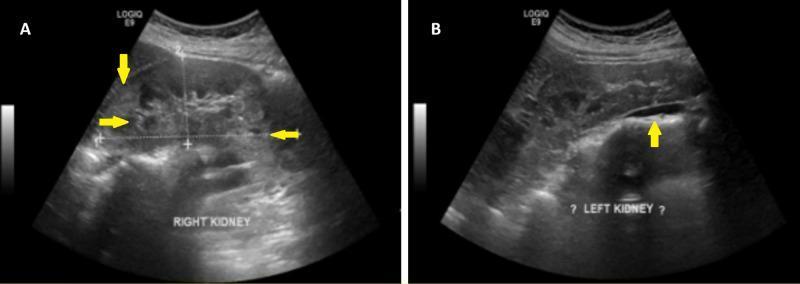
- Angiomyolipomas (AMLs): Most common renal finding. Multiple, bilateral. Composed of fat, smooth muscle, and abnormal blood vessels. Usually benign but prone to spontaneous hemorrhage (Wunderlich syndrome), especially >4 cm. Prophylactic embolization or surgery recommended for lesions >4 cm.
- Renal cysts: Present in 20–30%. Radiographically similar to ADPKD cysts but generally smaller burden.
- Renal cell carcinoma: Clearly elevated risk; requires regular imaging surveillance.
- Chronic kidney disease: Rare but may progress to end-stage renal failure. Urine sediment unremarkable; only mild proteinuria.
— Harrison's Principles of Internal Medicine 22E
Heart
- Cardiac rhabdomyomas: Most common cardiac tumor of childhood. Often detected on fetal echocardiography — may be the first sign of TSC. Can cause arrhythmias or outflow obstruction in neonates. Most spontaneously regress after infancy.
Lungs
- Lymphangioleiomyomatosis (LAM): Proliferation of smooth muscle-like cells along lymphatics, airways, and blood vessels. Predominantly affects women of reproductive age. Causes progressive dyspnea, recurrent pneumothorax, and chylothorax.
Eyes
- Retinal hamartomas (phakomas): Flat, translucent glial lesions or calcified "mulberry" nodules at the optic disc or peripheral retina. Usually asymptomatic.
Other
- Liver: Hepatic angiomyolipomas (usually asymptomatic)
- Pancreas/kidneys/liver: Cysts may be present at multiple sites
Pathology
The fundamental lesion is a hamartoma — a disorganized but benign proliferation of tissue elements native to the site. Hallmarks across tissue types:
- Balloon cells / giant cells: Large cells with abundant pale cytoplasm, eccentric nuclei, and features intermediate between neurons and glia
- Disorganized cortical lamination with loss of gray-white matter boundaries
- Calcification in SENs
The hamartin–tuberin complex normally acts as a GTPase-activating protein for RHEB, maintaining mTOR in an off state. Haploinsufficiency (and likely a "second hit" in some lesions) leads to mTOR overactivation, which drives increased cellular anabolism, dysregulated proliferation, and aberrant differentiation — producing hamartomas throughout the body.
— Robbins & Kumar Basic Pathology; Bradley and Daroff's Neurology in Clinical Practice
Management
| Domain | Intervention |
|---|---|
| Epilepsy | Antiepileptic drugs (often refractory); vigabatrin is first-line for TSC-associated infantile spasms; epilepsy surgery for refractory tuber-driven seizures; everolimus reduces seizure frequency |
| SEGA | Surgical resection for acute hydrocephalus; everolimus (mTOR inhibitor) shrinks SEGA and is used for non-surgical candidates or prophylaxis |
| Renal AMLs >4 cm | Selective arterial embolization; nephron-sparing surgery; everolimus (FDA-approved for TSC-associated renal AMLs) |
| LAM | Sirolimus or everolimus slow lung function decline (FDA-approved); lung transplant for end-stage |
| Facial angiofibromas | CO₂ or pulsed-dye laser; topical sirolimus (mTOR inhibitor) cream |
| Cardiac rhabdomyomas | Usually watchful waiting — spontaneous regression expected; treat arrhythmias if present |
| Surveillance | Annual brain MRI (SEGA monitoring), renal imaging every 1–3 years, echocardiography, ophthalmology, neuropsychiatric evaluation, pulmonary function (especially women) |
mTOR inhibitors (everolimus, sirolimus) are now first-line treatment across multiple TSC manifestations simultaneously — renal AMLs, SEGA, LAM, and epilepsy — making them the most broadly applicable therapy in TSC.
— Harrison's Principles of Internal Medicine 22E; Bradley and Daroff's Neurology in Clinical Practice
Key Points
- TSC is a neurocutaneous (phakomatosis) syndrome; skin findings are almost universal and usually the first diagnostic clue
- Driven by loss-of-function in TSC1 (hamartin) or TSC2 (tuberin) → mTOR overactivation
- TSC2 mutations cause more severe disease and are more common in sporadic cases
- The 2012 criteria formalize 11 major and 6 minor clinical features; a confirmed pathogenic mutation alone suffices for diagnosis
- mTOR inhibitors have transformed management, offering disease-modifying therapy across brain, kidney, and lung simultaneously
- TAND (neuropsychiatric manifestations) is a major source of lifetime morbidity and requires proactive assessment
This is a shared conversation. Sign in to Orris to start your own chat.