(Choice A) These arterial blood gas findings are consistent with polycythemia vera (PV), a condition characterized by an increase in erythrocyte count and thus hemoglobin (Hb) concentration. While this results in an increased arterial O2 content, O2 partial pressure and O2 saturation remain normal because these parameters mainly depend on the inhaled oxygen concentration, atmospheric pressure, and gas exchange in the lungs, none of which is affected by increased Hb concentration. Classical manifestations of PV include facial flushing and pruritus, neither of which is seen in this patient. Moreover, respiratory depression, which is seen here, would not be expected. (Choice B) These arterial blood gas findings are consistent with anemia, which is characterized by low serum concentrations of hemoglobin (Hb) and, as a consequence, reduced arterial O2 content. O2 partial pressure and O2 saturation remain normal because these parameters mainly depend on the inhaled oxygen concentration, atmospheric pressure, and gas exchange in the lungs, none of which is affected by low Hb concentration. Anemia can cause tachycardia, which is seen in this patient, but pallor rather than cyanosis would be expected. Moreover, chocolate-colored blood that does not change color when exposed to oxygen is not seen in anemia. (Choice C) Methemoglobinemia is caused by an abundance of methemoglobin, an altered form of hemoglobin in which ferrous iron (Fe2+) is oxidized to the ferric form (Fe3+). Since Fe3+ cannot bind oxygen, arterial O2 saturation and total O2 content decrease. This leads to functional anemia, despite normal hemoglobin concentrations. Patients with methemoglobinemia show clinical signs of cyanosis due to poor tissue oxygenation. O2 partial pressure (PaO2), which reflects the concentration of dissolved oxygen in plasma, is not affected. In patients prone to developing methemoglobinemia (e.g., patients with G6PD deficiency), oxidant drugs such as dapsone and topical anesthetic drugs should be avoided. Treatment consists of converting methemoglobin to hemoglobin by using reducing agents such as methylene blue and vitamin C. Although prolonged or chronic methemoglobinemia may lead to erythrocytosis or polycythemia, this patient has only been taking her medication for three days. See “Overview of factors that affect oxygenation” table. (Choice D) These arterial blood gas findings are consistent with hypoxemia (e.g., due to impaired gas exchange in the lungs). Impaired oxygenation of the blood decreases both the amount of oxygen dissolved in the plasma (↓ PaO2) and the amount of oxygen bound to hemoglobin (↓ SaO2). As a result, the total amount of oxygen transported by arterial blood (i.e., O2 content) will also be reduced. In states of hypoxemia, the hemoglobin concentration is initially unaffected, but may eventually increase due to stimulation of erythropoietin secretion. Hypoxemia can cause signs of cyanosis, which are seen here. However, unlike this patient's blood sample, deoxygenated blood changes color to bright red upon exposure to oxygen. (Choice E) These arterial blood gas findings are consistent with chronic carbon monoxide poisoning (e.g., from cigarette smoke). Since carbon monoxide binds to hemoglobin (Hb) with much higher affinity than oxygen (producing carboxyhemoglobin), it cannot easily be displaced. This resultsotrs
Why Each Choice is Correct or Incorrect
✅ Choice C — Methemoglobinemia (CORRECT)
| Parameter | Value | Reason |
|---|---|---|
| PaO₂ | Normal | Reflects O₂ dissolved in plasma — unaffected by Hb oxidation state |
| SaO₂ | ↓ | Measured by co-oximetry; methemoglobin cannot bind O₂ |
| Total O₂ content | ↓ | Because O₂ carrying capacity is reduced |
| Hb concentration | Normal | Red cell count intact |
- Chocolate-brown blood that does not turn bright red upon O₂ exposure (unlike deoxyhemoglobin, which brightens readily)
- Cyanosis disproportionate to PaO₂
- Tachycardia from compensatory response to functional anemia
- Exposure to an oxidant drug (e.g., dapsone, topical anesthetics, nitrates, primaquine) — the patient has been on a new medication for 3 days
❌ Choice A — Polycythemia Vera
- Increased Hb → increased O₂ content, but PaO₂ and SaO₂ remain normal
- Would cause facial flushing, pruritus — not seen here
- No chocolate-brown blood; no respiratory depression expected
❌ Choice B — Anemia
- Decreased Hb → decreased O₂ content, but PaO₂ and SaO₂ normal
- Would cause pallor, not cyanosis
- Chocolate-brown, oxygen-unresponsive blood is not a feature
❌ Choice D — Hypoxemia (impaired gas exchange)
- Would show ↓ PaO₂ AND ↓ SaO₂ (both reduced)
- Deoxygenated blood does turn bright red on O₂ exposure — distinguishing it from methemoglobinemia
- Normal PaO₂ in this patient rules this out
❌ Choice E — Chronic Carbon Monoxide Poisoning
- CO binds Hb with ~240× the affinity of O₂, forming carboxyhemoglobin (COHb)
- PaO₂ is normal (same as methemoglobinemia); SaO₂ by co-oximetry is reduced
- Key difference: CO poisoning causes cherry-red skin (not chocolate-brown blood) and occurs with chronic exposure (e.g., cigarette smoke, faulty heating)
The Distinguishing Clue: Chocolate-Brown Blood
"Methemoglobinemic blood is chocolate brown, and spectrophotometric examination of blood reveals the characteristic pigment. Arterial blood examination discloses a normal PO₂." — Fishman's Pulmonary Diseases and Disorders
can you make a HY compact notes how test makers want to tesr usmle step 1 compact notes easy question solving with concept grip of above asso. conditions and options filter and buzzwords
🧠 HY USMLE Step 1: Oxygen Transport Disorders
"ABG + Clinical Buzzword = Answer"
🔑 THE MASTER FRAMEWORK — Read This First
| Parameter | What it measures | Affected by Hb? |
|---|---|---|
| PaO₂ | O₂ dissolved in plasma | ❌ NO |
| SaO₂ | % Hb saturated with O₂ | ✅ YES |
| O₂ Content | Total O₂ in blood (= 1.36 × Hb × SaO₂ + 0.003 × PaO₂) | ✅ YES |
Core rule: PaO₂ depends on lungs + atmosphere only. SaO₂ and O₂ content depend on functional hemoglobin.
⚡ THE 5-CONDITION GRID
| Condition | PaO₂ | SaO₂ | O₂ Content | Hb conc. | Blood color | Cyanosis? |
|---|---|---|---|---|---|---|
| Polycythemia Vera | N | N | ↑ | ↑↑ | Red | No (flushing) |
| Anemia | N | N | ↓ | ↓↓ | Pale | No (pallor) |
| Methemoglobinemia | N | ↓ | ↓ | N | Chocolate-brown | Yes |
| Hypoxemia | ↓ | ↓ | ↓ | N (initially) | Dark → turns RED with O₂ | Yes |
| CO Poisoning | N | ↓ | ↓ | N | Cherry-red skin | Rare |
🎯 BUZZWORD FILTER — Crack the Stem in Seconds
🟣 Polycythemia Vera
Buzzwords: facial flushing • pruritus after hot shower • splenomegaly • JAK2 mutation • ↑Hct • thrombosis • erythromelalgia
- PaO₂ normal, SaO₂ normal, O₂ content HIGH
- They give you: plethoric face + itching after bath → PV
- Trap: may progress to AML or myelofibrosis
🔴 Anemia
Buzzwords: pallor • fatigue • tachycardia • koilonychia (Fe def) • glossitis • no cyanosis
- PaO₂ normal, SaO₂ normal, O₂ content LOW
- They give you: pale + tachycardia → Anemia
- Trap: never cyanosis, never chocolate blood
🟤 Methemoglobinemia ⭐ (HIGHEST YIELD)
Buzzwords: chocolate-brown blood • blood does NOT turn red with O₂ • cyanosis despite normal PaO₂ • oxidant drug • pulse ox reads ~85% regardless
- PaO₂ NORMAL, SaO₂ ↓, O₂ content ↓
- Mechanism: Fe²⁺ → Fe³⁺ (cannot carry O₂)
- Classic culprit drugs: dapsone • benzocaine/lidocaine (topical anesthetics) • nitrates/nitrites • primaquine • phenazopyridine • sulfonamides
- Treatment: Methylene blue (requires NADPH via G6PD)
- Trap: G6PD deficiency → methylene blue FAILS → use vitamin C instead
- Pulse ox paradox: reads falsely ~85% (SaO₂ looks "ok" but co-oximetry shows true ↓)
🔵 Hypoxemia
Buzzwords: low PaO₂ • ventilation-perfusion mismatch • blood turns BRIGHT RED with O₂ • cyanosis • ↑RR • may develop polycythemia over time (EPO stimulus)
- PaO₂ ↓, SaO₂ ↓, O₂ content ↓
- Key differentiator from methemoglobinemia: blood re-oxygenates (turns red) when exposed to air/O₂
- Causes: PE, ARDS, pneumonia, high altitude, hypoventilation
🟥 Carbon Monoxide Poisoning
Buzzwords: cherry-red skin (or normal) • headache • no cyanosis • normal PaO₂ • smoker or poorly ventilated space • pulse ox falsely NORMAL (reads COHb as OxyHb)
- PaO₂ NORMAL (same as methemoglobinemia!)
- SaO₂ ↓ by co-oximetry only
- Mechanism: CO binds Hb with 240× affinity of O₂ → left-shifts O-dissociation curve
- Treatment: 100% O₂ (or hyperbaric O₂ in severe cases)
- Trap vs. Methemoglobinemia: CO → cherry-red, no chocolate blood; Meth → chocolate blood, no cherry-red
🔬 THE TWO GREAT MIMICS (Most Tested Trap)
Normal PaO₂ + Cyanosis = Methemoglobinemia OR CO poisoning
Differentiator:
→ Chocolate-brown blood + oxidant drug = METHEMOGLOBINEMIA
→ Cherry-red skin + CO source = CO POISONING
→ Pulse ox 85% plateau = Methemoglobinemia
→ Pulse ox falsely normal = CO Poisoning
💊 DRUG → CONDITION MAP (USMLE Favorite)
| Drug given in stem | Condition triggered |
|---|---|
| Dapsone | Methemoglobinemia |
| Benzocaine / lidocaine (topical) | Methemoglobinemia |
| Nitrates (nitroglycerin, amyl nitrite) | Methemoglobinemia |
| Primaquine | Methemoglobinemia (+ hemolysis in G6PD) |
| Phenazopyridine | Methemoglobinemia |
| Carbon monoxide (smoke/car/heater) | CO poisoning |
| Erythropoietin / chronic hypoxia | Secondary polycythemia |
🧪 TREATMENT QUICK TABLE
| Condition | Treatment | Mechanism |
|---|---|---|
| Methemoglobinemia | Methylene blue (1st line) | Activates NADPH-MetHb reductase (needs G6PD!) |
| Methemoglobinemia in G6PD def. | Vitamin C (ascorbic acid) | Direct chemical reduction |
| CO Poisoning | 100% O₂ → hyperbaric O₂ | Displaces CO from Hb |
| PV | Phlebotomy + hydroxyurea | Reduces RBC mass |
| Anemia | Treat underlying cause | — |
🎲 HOW STEP 1 WRITES THE QUESTION
Stem gives: ABG (normal PaO₂) + cyanosis + chocolate blood + dapsone → Answer: Methemoglobinemia
Stem gives: topical anesthetic before endoscopy → cyanosis + normal PaO₂ → Answer: Oxidation of Fe²⁺ to Fe³⁺ in heme
Stem gives: methylene blue given → no improvement + G6PD deficiency noted → Answer: Methylene blue requires NADPH generated by G6PD — deficiency prevents reduction
CO vs. Methemoglobinemia → both normal PaO₂ → Key: blood color + co-oximetry + clinical context
Methemoglobinemia: pulse ox saturates at ~85% regardless of severity CO poisoning: pulse ox reads falsely normal
🗂 ONE-LINER MEMORY PEGS
| Condition | Memory Peg |
|---|---|
| PV | "Too many red cells, too much itch, JAK2 mutation" |
| Anemia | "Too few red cells, pale, tired, no cyanosis" |
| Methemoglobinemia | "Chocolate blood + normal PaO₂ + oxidant drug → blue patient" |
| Hypoxemia | "Low PaO₂, blood turns red with O₂, V/Q mismatch" |
| CO poisoning | "Cherry red, normal PaO₂, CO source, pulse ox lies" |
Bottom line for the exam: When you see cyanosis + normal PaO₂ — your reflex must be methemoglobinemia vs. CO. The blood color and drug history make the call. Everything else (anemia, PV, hypoxemia) cannot produce that ABG pattern.
Choice A) HMWK is a circulating plasma protein that is the precursor of the vasodilator bradykinin. During tissue injury, HMWK is activated by kallikrein, leading to an increase in vascular permeability, which facilitates cell migration to the site of injury. While kallikrein is a potent neutrophil chemotactic agent, HMWK is not known to act as a neutrophil chemokine and would not have played a role in the recruitment of neutrophils to the site of inflammation in this patient's lungs. (Choice B) Lactoferrin is a glycoprotein component of the innate immune system. It binds free iron with high affinity, thus limiting the amount of ions available for microbial metabolism. It also acts as an antimicrobial agent by hydrolyzing RNA of foreign bodies. It is stored in neutrophil granules and is released as an acute phase reactant. However, lactoferrin does not function as a neutrophil chemotactic agent and would not have played a role in the recruitment of neutrophils to the site of inflammation in this patient's lungs. (Choice C) IFN-γ is a cytokine involved in both innate and adaptive immune responses. Activation of macrophages increases the production of macrophage elastases, which damage the parenchymal structure of the lungs, leading to emphysema. However, macrophage elastases are inactivated by tissue inhibitors of metalloproteinases (TIMPs), not α1-antitrypsin, which inactivates neutrophil elastases. (Choice D) Nitric oxide (NO) is a gaseous free radical produced in some cells as a signaling molecule (produced by NO synthase) or as part of the immune response. Once activated, neutrophils secrete NO in an attempt to kill pathogens with the free radical. However, NO is not known to possess neutrophil chemotactic properties and would not have played a role in the recruitment of neutrophils to the site of inflammation in this patient's lungs. (Choice E) C4a is a product of the complement pathways. The function of the molecule is not well understood, though it is thought to induce calcium mobilization in cells. While C4a is structurally similar to the potent neutrophil chemotactic factor C5a, C4a is not known to possess neutrophil chemotactic properties and would not have played a role in the recruitment of neutrophils to the site of inflammation in this patient's lungs. (Choice F) Leukotriene B4, along with IL-8, kallikrein, C5a, and platelet-activating factor are all neutrophilic chemotactic agents. Recruitment and activation of neutrophils in the lungs lead to an increase in elastase production and damage to the parenchymal tissue. In cases of AATD, the unopposed enzymatic activity of the proteases destroys the architecture of whole pulmonary acini, leading to panacinar emphysema. (Choice G) TXA2 is an arachidonic acid derivative produced by activated platelets. It plays an important role during tissue injury and inflammation as a potent vasoconstrictor. However, TXA2 is not a known neutrophil chemokine and would not have played a role in the recruitment of neutrophils to the site of inflammation in this patient's lungs. Make USMLE HY ultra compact notes fast revision
⚡ HY USMLE Step 1: Neutrophil Chemotaxis & AATD — Ultra Compact Notes
🎯 THE CORE CONCEPT THIS QUESTION TESTS
"Which mediator RECRUITS neutrophils to a site of inflammation?" Specifically in the context of Alpha-1 Antitrypsin Deficiency (AATD) → Panacinar Emphysema
🔑 MASTER LIST: Neutrophil Chemotactic Agents (MEMORIZE ALL)
IL-8 • LTB4 • C5a • Kallikrein • PAF (Platelet-Activating Factor) • fMLP (bacterial)
Mnemonic: "I Like C5, KP Fights" IL-8 | LTB4 | C5a | Kallikrein | PAF | fMLP
❌ THE DECOYS — NOT Neutrophil Chemokines (What USMLE uses to trap you)
| Molecule | What it DOES do | NOT a chemokine? |
|---|---|---|
| HMWK | Precursor of bradykinin → ↑vascular permeability | ❌ Not chemotactic |
| Lactoferrin | Binds free iron → antimicrobial; stored in neutrophil granules | ❌ Not chemotactic |
| Nitric Oxide (NO) | Kills pathogens (free radical); made by NO synthase | ❌ Not chemotactic |
| C4a | Calcium mobilization; structurally similar to C5a | ❌ Not chemotactic |
| TXA2 | Potent vasoconstrictor; from activated platelets | ❌ Not chemotactic |
| IFN-γ | Activates macrophages → macrophage elastase → emphysema | ❌ Not chemotactic |
🔬 THE AATD PATHWAY (Full Chain — Step 1 Blueprint)
AATD (chromosome 14, PI*ZZ genotype)
↓
↓ α1-antitrypsin (serine protease inhibitor)
↓
Neutrophils recruited by: LTB4, IL-8, C5a, Kallikrein, PAF
↓
↑ Neutrophil elastase — UNOPPOSED (no AAT to inhibit it)
↓
Destroys entire pulmonary acinus (alveolar walls + capillaries)
↓
PANACINAR emphysema (lower lobe predominant)
Trap distinction:
- AAT inhibits → neutrophil elastase
- TIMPs inhibit → macrophage metalloproteinases (MMP)
- IFN-γ activates macrophages → MMP pathway → separate emphysema mechanism
💥 BUZZWORD → ANSWER MAP
| Stem says... | Think... |
|---|---|
| Panacinar emphysema, lower lobes, young non-smoker | AATD |
| Liver disease (cirrhosis) in same patient | AATD (misfolded Z protein accumulates in hepatocytes) |
| "Neutrophil chemotactic factor" in MCQ | LTB4, IL-8, C5a, kallikrein, PAF |
| "Unopposed elastase activity" | AATD |
| "Macrophage elastase inhibited by..." | TIMPs (NOT AAT) |
| "Neutrophil elastase inhibited by..." | α1-antitrypsin |
| Free iron binding, antimicrobial | Lactoferrin |
| Bradykinin precursor, ↑permeability | HMWK |
| Vasoconstrictor, platelet-derived | TXA2 |
| Gaseous free radical, kills bacteria | NO |
| Calcium mobilization, complement | C4a (not C5a!) |
🧪 COMPLEMENT CHEMOTAXIS — DON'T CONFUSE
C3a → Anaphylatoxin (mast cell degranulation, NOT chemotaxis)
C4a → Weak anaphylatoxin, calcium mobilization — NOT chemotactic
C5a → BOTH anaphylatoxin AND potent neutrophil CHEMOTAXIN ✅
Rule: Only C5a from complement is a neutrophil chemokine. C3a and C4a are NOT.
🏗 ARACHIDONIC ACID DERIVATIVES — QUICK SORT
| Derivative | Made by | Function |
|---|---|---|
| LTB4 | Neutrophils/mast cells | Neutrophil chemotaxis ✅ |
| LTC4, LTD4, LTE4 | Mast cells | Bronchoconstriction, ↑mucus |
| TXA2 | Platelets | Vasoconstriction, platelet aggregation |
| PGI2 (prostacyclin) | Endothelium | Vasodilation, ↓platelet aggregation |
| PGE2 | Many cells | Fever, vasodilation, pain |
LTB4 = the ONLY arachidonic acid derivative that is a neutrophil chemokine
🎲 HOW STEP 1 WRITES THIS QUESTION
AATD patient, panacinar emphysema → neutrophils in lung Correct answer: LTB4 or IL-8 or C5a
Answer: α1-antitrypsin (not TIMPs — those are for macrophage MMPs)
Answer: C5a only — C4a is structurally similar but NOT chemotactic (classic USMLE trap)
Answer: Misfolded Z-protein (Glu→Lys substitution) accumulates in hepatocyte ER → cirrhosis; it's a loss-of-function AND toxic gain-of-function
AATD → Panacinar, lower lobe Smoking → Centrilobular (centriacinar), upper lobe
🗂 ONE-LINE MEMORY PEGS
| Molecule | Peg |
|---|---|
| LTB4 | "B4 the neutrophil arrives" — B4 = chemotaxis |
| C5a | "C5 drives neutrophils alive" |
| IL-8 | "IL-8, neutrophils don't wait" |
| HMWK | "High Molecular Weight Kininogen → bradyKinin → NOT a chemokine" |
| TXA2 | "TXA2 constricts, never recruits" |
| Lactoferrin | "Iron thief in neutrophil granules — not a GPS signal" |
| C4a vs C5a | "C4a is the fake C5a — same shape, no game" |
✅ BOTTOM LINE FOR THE EXAM
When the stem describes panacinar emphysema + young patient + liver disease → AATD The neutrophils got there via LTB4 / IL-8 / C5a / kallikrein / PAF Once there, their unopposed elastase (no AAT) destroys acini Treatment: IV pooled AAT (augmentation therapy) or lung transplant AATD liver tx = most common metabolic cause of liver transplant in children
what is this aatd disease
Alpha-1 Antitrypsin Deficiency (AATD) — Complete Explanation
🧬 What is Alpha-1 Antitrypsin (AAT)?
Neutrophils arrive at inflammation site
↓
Release elastase (a powerful enzyme that digests proteins)
↓
AAT immediately blocks elastase → stops collateral lung damage
↓
Without AAT → elastase runs unchecked → destroys lung tissue
🧬 The Genetic Defect
| Genotype | AAT level | Risk |
|---|---|---|
| PiMM | Normal | No disease |
| PiMZ | ~60% normal | Mild risk |
| PiZZ | ~10–15% normal | Severe disease |
🫁 What Happens in the LUNGS
Inflammation (even minor) → neutrophils recruited to lung
↓
Neutrophil elastase released
↓
NO AAT to stop it → elastase destroys entire pulmonary acinus
(respiratory bronchiole + alveolar ducts + alveoli — ALL destroyed)
↓
PANACINAR EMPHYSEMA — lower lobe predominant
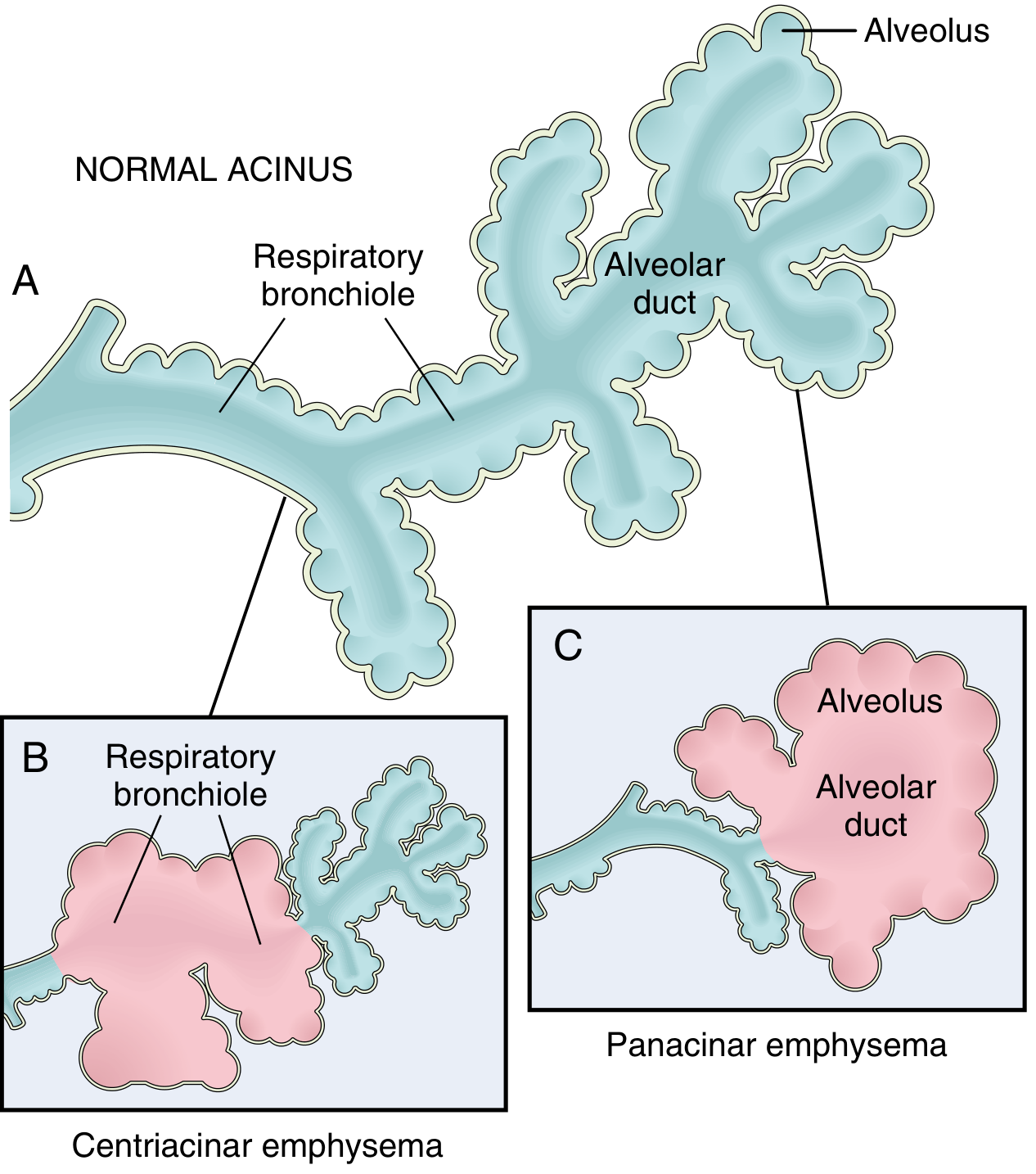
🫀 What Happens in the LIVER
Misfolded Z-AAT → stuck in ER → PAS-positive, diastase-resistant globules in hepatocytes
↓
ER stress → hepatocyte death → fibrosis → CIRRHOSIS
Key fact: The liver disease is caused by protein ACCUMULATION (toxic). The lung disease is caused by protein ABSENCE (loss of function). Same gene, two completely different mechanisms.
👤 Who Gets It & When Do They Present?
| Feature | Detail |
|---|---|
| Age of presentation | Lung: 30s–40s (earlier if smoker). Liver: infancy or adulthood |
| Ethnicity | Northern European descent (most common) |
| Smoking effect | Dramatically accelerates emphysema — smoking ↑ neutrophil influx AND ↑ oxidative inactivation of remaining AAT |
| ~1% of all emphysema patients have AATD |
🩺 Clinical Features
- Progressive dyspnea (breathlessness)
- Chronic obstructive pattern on PFTs (↓ FEV1/FVC)
- Hyperinflated lungs on CXR — lower lobe hyperinflation (opposite of smoking emphysema which is upper lobe)
- Barrel chest, pursed-lip breathing
- Neonatal cholestasis / hepatomegaly in infants
- Cirrhosis in adults
- Hepatocellular carcinoma (long-term)
- Most common metabolic disease requiring liver transplant in children
🔬 Pathology Findings
| Test | Finding |
|---|---|
| Lung histology | Panacinar emphysema — enlarged airspaces throughout entire acinus, lower lobes |
| Liver histology | PAS-positive, diastase-resistant globules in hepatocytes (misfolded Z-AAT protein) |
| Serum | Very low AAT levels |
| Genotyping | PiZZ confirmed by isoelectric focusing or PCR |
💊 Treatment
| Target | Treatment |
|---|---|
| Lung disease | IV pooled human AAT augmentation therapy (replaces the missing protein) — slows progression |
| Severe lung disease | Lung transplant |
| Severe liver disease | Liver transplant (cures both liver AND lung disease since transplanted liver makes normal AAT) |
| All patients | Smoking cessation — most important modifiable factor |
⚡ USMLE Summary in 5 Lines
- Gene: Pi locus, chromosome 14 — PiZZ = disease
- Mechanism: Misfolded Z-protein → stuck in liver ER (liver damage) + absent in blood (no elastase inhibition → lung damage)
- Lung: Panacinar emphysema, lower lobes, young non-smoker or mild smoker
- Liver: PAS+ diastase-resistant globules, cirrhosis, #1 metabolic cause of pediatric liver transplant
- Rx: AAT infusions for lung; liver transplant cures both organs
So usually in a question stem, they're asking that the patient came with a chief complaint of cough and dyspnea and smoking history he had. So x-ray taken and it's showing that the diaphragm is flat and the lower lobes shows uh increased translucency or something like that. And in the summary, they say that there is an increased isoform of LSD which is present and alpha 1 antitrypsin deficient. The patient is for this uh isoform of LSD which is found. Which organism is responsible for this LSD formation and what are the stimulating factors for this uh neutrophilic elastase? Then we have to look up about neutrophilic hematologic agents. There they given all these options, how smartly they tested. That I want as a classical clinical vignette kind of question, uh clinical vignette uh type of uh uh making so that I can write in my notes for easy reference.
🏥 CLASSICAL USMLE CLINICAL VIGNETTE — AATD
📋 THE VIGNETTE
A 42-year-old man presents with a 6-month history of progressive dyspnea and productive cough. He has a 30-pack-year smoking history. He denies fever or recent travel. On physical exam, he has a barrel chest, decreased breath sounds bilaterally, and prolonged expiration with pursed-lip breathing. There is no peripheral edema.CXR shows: Hyperinflated lungs, flattened diaphragm, and increased translucency predominantly in the lower lobes.PFTs: FEV1/FVC = 0.55 (obstructive pattern), markedly reduced DLCO.Labs: Serum protein electrophoresis shows a decreased alpha-1 globulin peak. Genotyping confirms PiZZ phenotype. A sputum culture reveals gram-negative organisms whose lipopolysaccharide (LPS/endotoxin) is identified as the triggering isoform responsible for the current exacerbation.
❓ QUESTION
Which of the following mediators was MOST responsible for recruiting neutrophils to the lung parenchyma, thereby initiating the elastase-mediated destruction seen in this patient?A. High-molecular-weight kininogen (HMWK) B. Lactoferrin C. IFN-γ D. Nitric oxide (NO) E. C4a F. Leukotriene B4 (LTB4) G. Thromboxane A2 (TXA2)
🔑 HOW TO DECODE THIS VIGNETTE IN 30 SECONDS
Lower lobe hyperinflation + flattened diaphragm
↓
Panacinar emphysema pattern → think AATD immediately
Decreased alpha-1 globulin peak + PiZZ
↓
Confirmed AATD
Gram-negative bacteria → LPS (endotoxin) released
↓
LPS activates macrophages & mast cells
↓
LTB4 + IL-8 + C5a + PAF secreted → NEUTROPHIL CHEMOTAXIS
↓
Neutrophils flood the lung → release ELASTASE
↓
No AAT to stop it → destroys entire acinus → PANACINAR EMPHYSEMA
🦠 THE ORGANISM & LPS ANGLE
| Question asked | Answer |
|---|---|
| What organism produces LPS? | Gram-negative bacteria (Pseudomonas, H. influenzae, Klebsiella — common in COPD exacerbations) |
| What is LPS? | Lipopolysaccharide = component of gram-negative outer membrane = powerful endotoxin |
| What does LPS do to neutrophils? | Directly activates neutrophils via TLR-4 (Toll-like receptor 4) AND stimulates macrophages to produce LTB4, IL-8, C5a → massive neutrophil recruitment |
| Why is LPS especially dangerous in AATD? | Normal lungs handle neutrophil elastase via AAT. In AATD, LPS-triggered neutrophil surge is UNOPPOSED → catastrophic acinar destruction |
⚡ STIMULATORS OF NEUTROPHIL ELASTASE RELEASE
This is what triggers neutrophils to dump their elastase:
| Stimulator | Source | Mechanism |
|---|---|---|
| LPS (endotoxin) | Gram-negative bacteria outer membrane | TLR-4 → NF-κB → neutrophil degranulation |
| Cigarette smoke | Direct oxidant + recruits neutrophils | Oxidizes AAT (inactivates it) + recruits MORE neutrophils |
| IL-8 | Macrophages, epithelial cells | Chemotaxis + activation |
| C5a | Complement activation | Chemotaxis + degranulation |
| LTB4 | Mast cells, macrophages | Chemotaxis + primes neutrophils |
| fMLP | Formyl-Met-Leu-Phe — bacterial peptide | Direct neutrophil activator via G-protein receptor |
| PAF | Platelets, endothelium | Priming + activation |
Smoking double-hit in AATD:
- Directly recruits more neutrophils → more elastase
- Oxidizes AAT → inactivates what little AAT remains → This is why smokers with PiZZ develop emphysema decades earlier
🎯 OPTION-BY-OPTION ELIMINATION LOGIC
| Option | What it does | Is it chemotactic? | Eliminate because... |
|---|---|---|---|
| A. HMWK | Bradykinin precursor → ↑vascular permeability | ❌ | Permeability ≠ chemotaxis |
| B. Lactoferrin | Binds iron → antimicrobial; stored in neutrophil granules | ❌ | Released BY neutrophils, doesn't recruit them |
| C. IFN-γ | Activates macrophages → macrophage elastase | ❌ | Macrophage elastase inhibited by TIMPs, not AAT |
| D. NO | Free radical, kills pathogens | ❌ | Signaling molecule, not a chemokine |
| E. C4a | Weak anaphylatoxin, Ca²⁺ mobilization | ❌ | Looks like C5a but NOT chemotactic |
| ✅ F. LTB4 | Neutrophil chemotaxis + activation | ✅ | Correct — classic neutrophil chemokine |
| G. TXA2 | Vasoconstriction, platelet aggregation | ❌ | Vascular function only |
🔬 THE TRAP IN THIS QUESTION
C4a vs C5a — the most common wrong answer
- Both from complement cascade
- C4a looks structurally similar to C5a
- Only C5a is chemotactic — C4a is NOT
- USMLE loves giving C4a as a distractor when the answer is a different chemokine
Lactoferrin trap — students think "it's in neutrophils so it recruits them"
- Wrong direction — lactoferrin is stored IN neutrophils and released AFTER they arrive
- It does NOT call neutrophils to the scene
📌 WRITE THIS IN YOUR NOTES — ONE BOX
╔══════════════════════════════════════════════════════════╗
║ AATD VIGNETTE — INSTANT RECOGNITION ║
╠══════════════════════════════════════════════════════════╣
║ BUZZWORDS: Lower lobe hyperinflation + flattened ║
║ diaphragm + young/mild smoker + ↓alpha-1 globulin peak ║
║ → PANACINAR emphysema → AATD (PiZZ, Chr 14) ║
╠══════════════════════════════════════════════════════════╣
║ ORGANISM → Gram-negative bacteria → LPS (endotoxin) ║
║ LPS via TLR-4 → macrophage activation ║
╠══════════════════════════════════════════════════════════╣
║ NEUTROPHIL CHEMOKINES (only these): ║
║ LTB4 • IL-8 • C5a • Kallikrein • PAF • fMLP ║
╠══════════════════════════════════════════════════════════╣
║ ELASTASE STIMULATORS: LPS • Smoking • C5a • fMLP ║
╠══════════════════════════════════════════════════════════╣
║ AAT inhibits → NEUTROPHIL elastase (chr 14, Pi locus) ║
║ TIMPs inhibit → MACROPHAGE metalloproteinases ║
╠══════════════════════════════════════════════════════════╣
║ TX: AAT augmentation IV (lung) | Liver Tx (both organs) ║
╚══════════════════════════════════════════════════════════╝
it was not LPS they said some isoform of elastase fancy way of saying neutrophil elasestase