cardiomyopathy
Reading File
Finding Sources
Searching PubMed
"cardiomyopathy"[MeSH Terms] AND ("classification" OR "management")
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Loading Image
Reading File
Finding Sources
Loading Image
Reading File
I now have comprehensive content from multiple authoritative sources. Let me compile the full answer.
Cardiomyopathy
Cardiomyopathies are intrinsic diseases of the cardiac muscle - distinct from heart disease secondary to coronary artery disease, hypertension, valvular abnormalities, or congenital defects. They are classified into three major pathophysiologic patterns based on functional impairment.
Classification Overview
| Type | LVEF | Primary Defect | Dominant Mechanism |
|---|---|---|---|
| Dilated (DCM) | <40% | Systolic (contractile) dysfunction | Impaired contractility |
| Hypertrophic (HCM) | 50-80% | Diastolic (relaxation) dysfunction | Impaired compliance |
| Restrictive (RCM) | Near normal | Diastolic dysfunction | Stiff, noncompliant walls |
DCM accounts for ~90% of cardiomyopathy cases; restrictive is the least common.
(Robbins & Kumar Basic Pathology, Table 9.5)
1. Dilated Cardiomyopathy (DCM)
Pathogenesis & Etiology
DCM is characterized by progressive cardiac dilation and contractile (systolic) dysfunction, usually with concurrent hypertrophy. At diagnosis it has typically progressed to end-stage heart failure with poor myocardial contractility. Key causes include:
- Genetic (20-50% of cases): Over 50 causal genes identified, predominantly autosomal dominant. Most commonly involves loss-of-function mutations in cytoskeletal or sarcomere-linking proteins:
- Titin (TTN) - the most common genetic cause; titin is key for sarcomeric force generation; truncating variants account for up to 20% of all DCM
- β-myosin heavy chain, α-myosin heavy chain, troponin T - also implicated
- Dystrophin (DMD gene, X-linked) - Duchenne and Becker muscular dystrophy
- Desmin - connects nuclear/plasma membranes, Z-lines, and intercalated disks; impairs both cardiac and skeletal muscle
- Nuclear lamins A and C - associated with conduction defects
- Viral infection: Coxsackievirus B, adenovirus, enteroviruses; increasingly parvovirus B19 and human herpesvirus 6. Viral nucleic acid footprints can persist into the late-stage DCM heart
- Alcohol/toxins: Ethanol and acetaldehyde have direct cardiotoxic effects; chronic alcohol use disorder is strongly associated with DCM
- Peripartum: DCM developing in the last trimester or within months of delivery
- Anthracyclines (e.g., doxorubicin): Dose-dependent cardiotoxicity
- Hemochromatosis, sarcoidosis, chronic anemia
- Idiopathic: A substantial proportion
Familial clustering is present in 30-40% of DCM cases; monogenic etiologies identifiable in ~25%.
(Harrison's 22E, p. 2052; Robbins & Kumar Basic Pathology, pp. 371-372)
Gross & Histologic Pathology
- Gross: Four-chamber dilation and hypertrophy; flabby, poorly contractile walls; mural thrombi (especially at LV apex). Heart weight often exceeds 800 g (upper limit of normal ~360 g). The LV wall appears thinned despite significant overall hypertrophy.
- Histology: Myocyte hypertrophy with interstitial fibrosis (nonspecific pattern); no diagnostic features on light microscopy at end-stage.
Dilated cardiomyopathy - gross specimen (heart removed at transplantation):
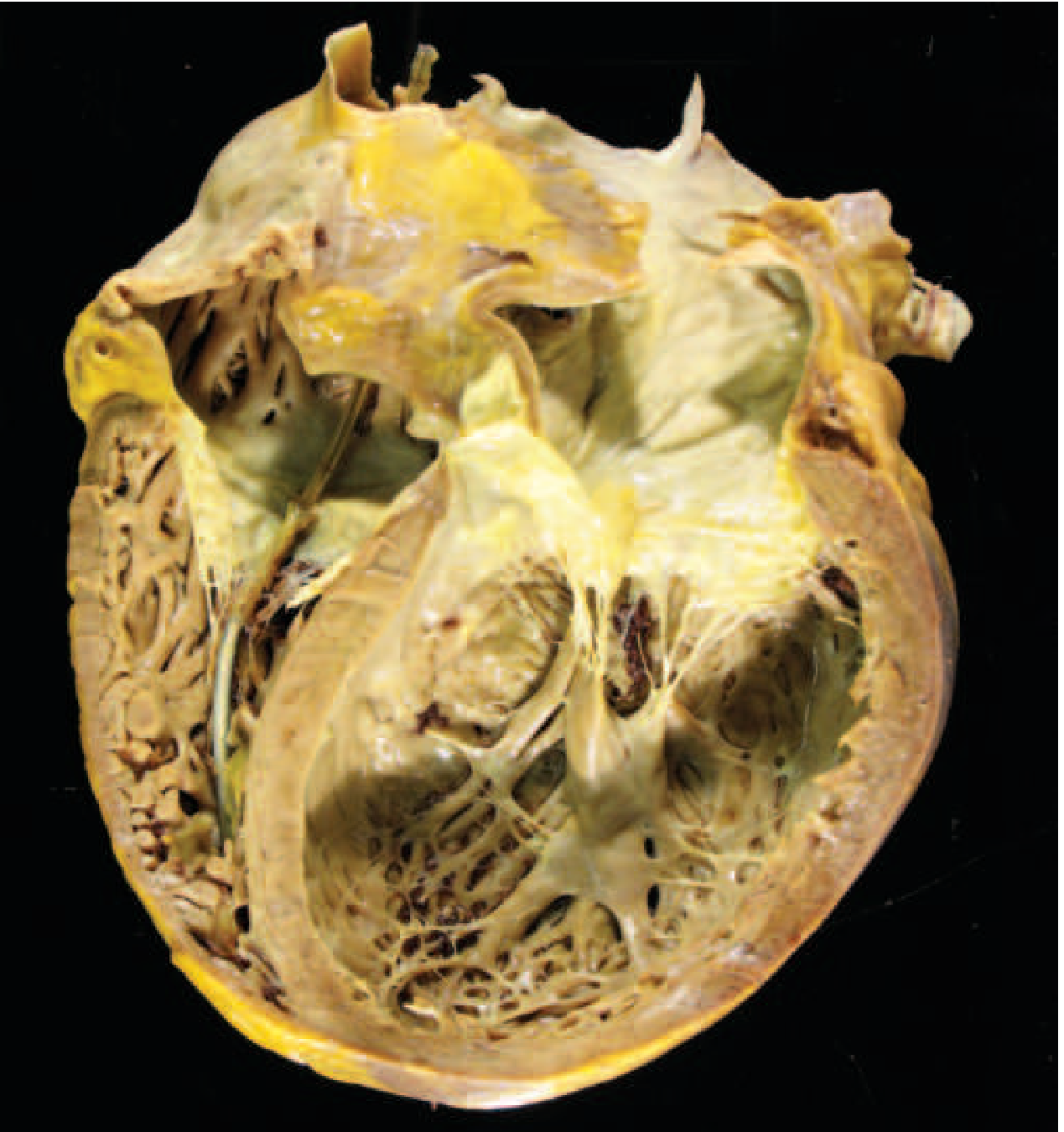
Massive left and right ventricular dilation with thinned but hypertrophied walls; defibrillator lead visible through tricuspid valve. Heart weighs >800 g. (Harrison's 22E, Fig. 267-6)
Clinical Features
Exertional dyspnea, fatigue, pulmonary congestion, peripheral edema, arrhythmias. High risk for embolic events (LV thrombus). Prognosis is poor without treatment; some cases (particularly viral-related) may partially recover.
2. Hypertrophic Cardiomyopathy (HCM)
Definition
HCM is defined as unexplained LV hypertrophy in the absence of abnormal loading conditions (valve disease, hypertension, congenital defects) sufficient to account for the degree of hypertrophy. Prevalence 0.2-0.5% worldwide, all racial groups. (Goldman-Cecil Medicine)
Pathogenesis
- Virtually always genetic: autosomal dominant gain-of-function mutations in sarcomeric contractile proteins
-
1,400 mutations identified in >9 genes; the three most common - MYH7 (β-myosin heavy chain), MYBPC3 (myosin-binding protein C), TNNT2 (troponin T) - account for 70-80% of all HCM cases
- The gain-of-function mutations cause myocyte hypercontractility, increased energy demand, and net negative energy balance, ultimately leading to pathological hypertrophy and fibrosis
- Contrast with DCM: the same gene (e.g., β-myosin) can cause either disorder depending on whether the mutation is gain-of-function (HCM) or loss-of-function (DCM)
(Robbins & Kumar Basic Pathology, pp. 373-374)
Gross & Histologic Pathology
- Gross: Massive myocardial hypertrophy without ventricular dilation
- 90% of cases: asymmetric septal hypertrophy - disproportionate thickening of the interventricular septum relative to the LV free wall
- 10% of cases: concentric hypertrophy
- On longitudinal section, LV cavity takes a "banana-shaped" configuration
- Fibrous endocardial plaque in the LV outflow tract (from contact of anterior mitral leaflet with septum during systole)
- Systolic anterior motion (SAM) of the mitral valve causes variable LV outflow tract obstruction in ~one-third of cases
- Histology: Extreme myocyte hypertrophy, haphazard myocyte (and myofiber) disarray (the hallmark), and interstitial fibrosis
HCM with asymmetric septal hypertrophy:
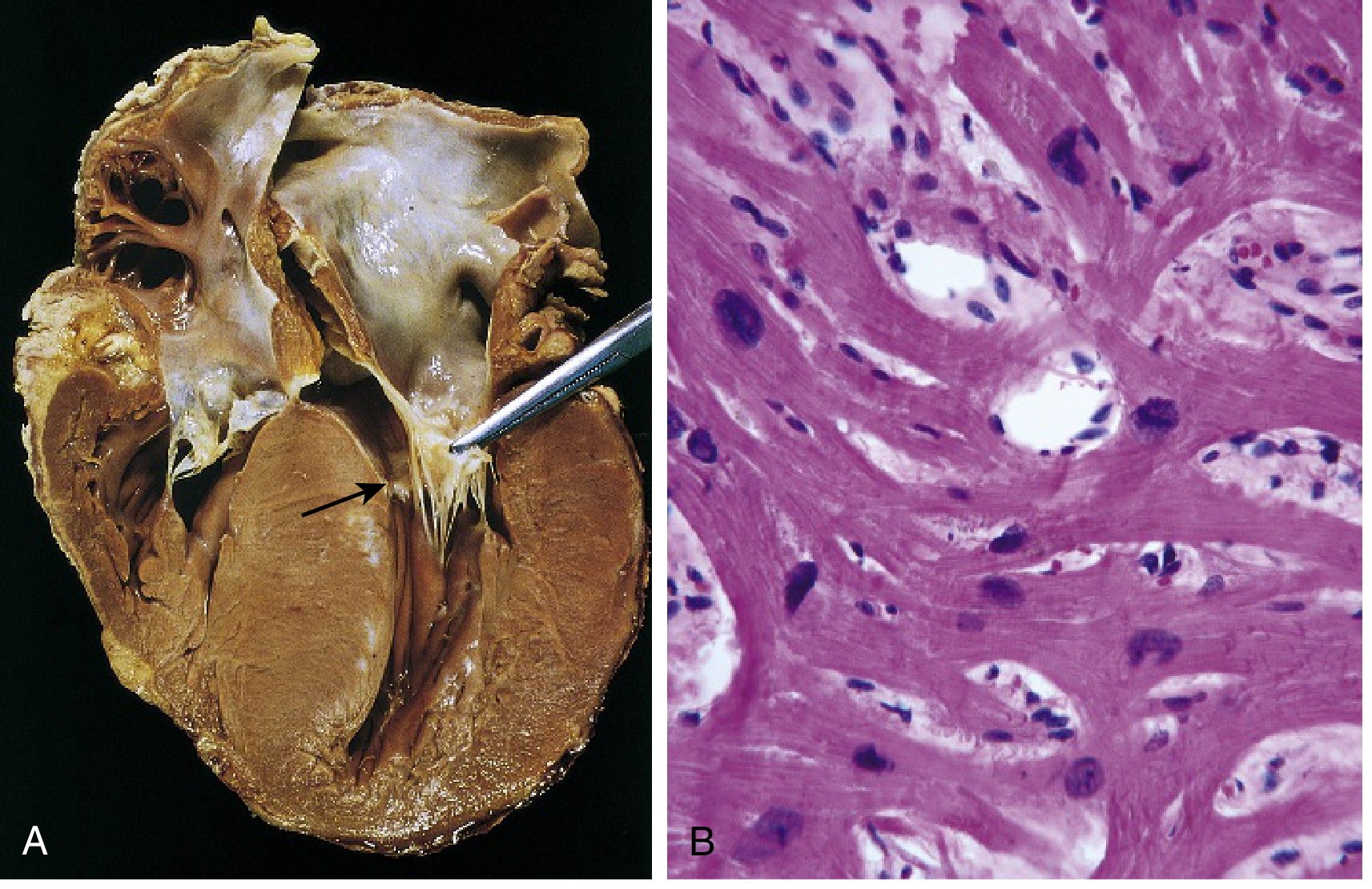
(A) Septal muscle bulges into LV outflow tract, creating a "banana-shaped" lumen; LA is enlarged; anterior mitral leaflet pulled away to show fibrous endocardial plaque (arrow). (B) Histology showing disarray, extreme hypertrophy, and interstitial fibrosis. (Robbins & Kumar Basic Pathology, Fig. 9.27)
Clinical Features
- Typically manifests during the postpubertal growth spurt, but can present at any age
- Diastolic dysfunction is the fundamental problem: the stiff, poorly relaxing LV limits filling, reducing stroke volume
- Symptoms: exertional dyspnea, harsh systolic ejection murmur, angina (even without CAD - caused by massive hypertrophy + high LV pressure + compromised intramural arterial flow)
- Complications:
- Atrial fibrillation with mural thrombus formation
- Sudden cardiac death from ventricular fibrillation - HCM is the most common cause of sudden cardiac death in athletes under 35 (accounting for ~1/3 of all such deaths)
- Infective endocarditis of the mitral valve
- Progressive CHF
- Treatment:
- Beta-blockers, calcium channel blockers (promote ventricular relaxation)
- Partial surgical septal myectomy
- Alcohol septal ablation (controlled therapeutic infarction of septal muscle via intracoronary alcohol injection)
- ICD for high-risk patients
(Robbins & Kumar Basic Pathology, pp. 373-374)
3. Restrictive Cardiomyopathy (RCM)
RCM is characterized by a decrease in ventricular compliance - the wall is abnormally stiff - resulting in impaired ventricular filling during diastole despite relatively preserved systolic function.
Gross morphology: Ventricles are approximately normal size (not dilated), myocardium is firm, but both atria are typically dilated due to chronically elevated ventricular filling pressures. Microscopy shows variable interstitial fibrosis.
Causes
| Cause | Key Feature |
|---|---|
| Cardiac amyloidosis | Deposition of transthyretin (TTR) or immunoglobulin light chains; TTR mutation in 4% of African Americans increases risk fourfold; AL amyloid light chains also directly cardiotoxic |
| Endomyocardial fibrosis | Most common RCM worldwide; children/young adults in Africa + tropical regions; diffuse fibrosis of ventricular endocardium/subendocardium, often involving tricuspid and mitral valves; linked to nutritional deficiencies/helminthic infections |
| Loeffler endomyocarditis | Peripheral hypereosinophilia and eosinophilic infiltrates; eosinophil granule products (especially major basic protein) cause endocardial/myocardial necrosis → scarring → mural thrombus → organization |
| Radiation fibrosis | Post-chest radiation |
| Sarcoidosis | Granulomatous infiltration |
| Storage diseases | Mucopolysaccharidoses, sphingolipidoses |
| Idiopathic |
(Robbins & Kumar Basic Pathology, pp. 374-375)
4. Arrhythmogenic Cardiomyopathy (ACM / ARVC)
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a separate genetic entity characterized by:
- Progressive replacement of the RV myocardium by fibro-fatty tissue
- The RV is markedly dilated; the LV may also be involved to a lesser extent
- Results in ventricular arrhythmias and sudden cardiac death, particularly in young athletes
Gross: RV markedly dilated with focal, almost transmural replacement of the free wall by adipose tissue and fibrosis.
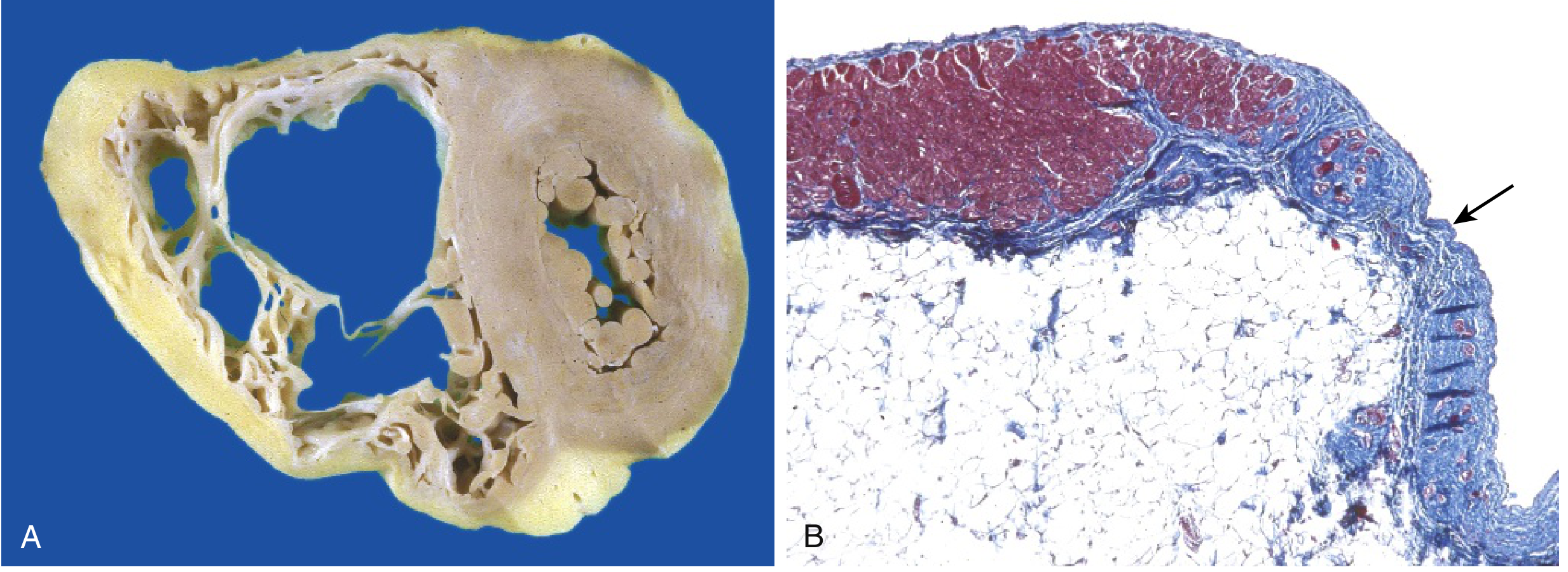
(A) RV markedly dilated with fibro-fatty replacement of free wall. (B) Masson trichrome showing RV myocardium (red) replaced by fibrosis (blue) and fat (arrow). (Robbins & Kumar Basic Pathology, Fig. 9.26)
The genetic basis of ACM involves mutations in desmosomal proteins (e.g., plakophilin-2, desmoplakin, desmoglein-2), which are critical for cell-cell adhesion in cardiomyocytes. (Harrison's 22E, p. 2052)
Key Comparison Table
| Feature | DCM | HCM | RCM | ACM |
|---|---|---|---|---|
| LV size | Dilated | Normal-enlarged | Normal/small | RV dilated |
| Wall thickness | Thin (relative) | Thick (massive) | Variable | RV thin |
| Systolic function | Severely impaired | Preserved | Preserved | RV impaired |
| Diastolic function | Secondarily impaired | Primarily impaired | Severely impaired | - |
| Key genetic lesion | TTN, dystrophin, desmin (loss-of-function) | MYH7, MYBPC3, TNNT2 (gain-of-function) | TTR, SCN5A | Desmosomal genes |
| Histology | Myocyte hypertrophy + interstitial fibrosis | Myocyte disarray + fibrosis | Fibrosis + infiltration | Fibro-fatty replacement |
| SCD risk | Moderate | High (young athletes) | Low-moderate | High (young athletes) |
Sources: Robbins & Kumar Basic Pathology (9th ed.); Robbins, Cotran & Kumar Pathologic Basis of Disease; Harrison's Principles of Internal Medicine, 22E; Goldman-Cecil Medicine; Robbins & Kumar Basic Pathology, Table 9.5
this elincludes all new guidelines , and everything about cardiomyopathy?
Asking for Preferences
Finding Sources
Finding Sources
Searching PubMed
"cardiomyopathy" AND "guidelines"
Searching the Web
ACC AHA ESC cardiomyopathy guidelines 2023 2024 HCM DCM ARVC updates
Reading Article
PMID: 38718139
Reading Article
PMID: 39674807
Reading Article
PMID: 41171219
Finding Sources
Reading Web Page
https://pmc.ncbi.nlm.nih.gov/articles/PMC12516781
Reading File
Reading File
Reading File
Searching PubMed
"dilated cardiomyopathy" OR "arrhythmogenic cardiomyopathy" AND "ESC guidelines"
Reading File
Reading File
Searching PubMed
"arrhythmogenic cardiomyopathy" AND "ESC" AND "management"
Searching the Web
2023 ESC guidelines cardiomyopathy DCM ARVC new classification NDLVC key recommendations
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading Web Page
https://www.revespcardiol.org/en-comments-on-the--esc-guidel…
Finding Sources
Here is the complete, exam-ready deep-dive on cardiomyopathy, incorporating the 2023 ESC Guidelines, 2024 AHA/ACC HCM Guidelines, 2025 ACC ATTR guidance, and all major textbook content.
CARDIOMYOPATHY - Complete Medical Student Guide
Updated with 2023 ESC, 2024 ACC/AHA, and 2025-2026 Guidance
PART 1: DEFINITION & CLASSIFICATION
Cardiomyopathies are primary diseases of the myocardium not explained by coronary artery disease, hypertension, valvular disease, or congenital heart disease. Prevalence: ~1 in 250 adults.
2023 ESC Classification (NEW - Replaces Older Systems)
The 2023 ESC Guidelines on Cardiomyopathies introduced a revised classification centered on morphological and functional phenotypes:
| Type | Abbreviation | Core Feature |
|---|---|---|
| Hypertrophic cardiomyopathy | HCM | LV hypertrophy without loading cause |
| Dilated cardiomyopathy | DCM | LV/biventricular dilation + systolic dysfunction |
| Non-dilated left ventricular cardiomyopathy | NDLVC | NEW: non-ischemic LGE or fatty replacement in LV, or isolated LV hypokinesia WITHOUT dilation |
| Arrhythmogenic right ventricular cardiomyopathy | ARVC | RV fibrofatty replacement + arrhythmias |
| Restrictive cardiomyopathy | RCM | Stiff, noncompliant ventricles, preserved size |
NDLVC is the key new entity - it encompasses conditions previously called left ventricular arrhythmogenic cardiomyopathy (LVAC) and non-compaction cardiomyopathy (NCC). It requires CMR for diagnosis.
(2023 ESC Guidelines for the Management of Cardiomyopathies, Eur Heart J 44:3503-3626)
PART 2: DILATED CARDIOMYOPATHY (DCM)
Definition & Epidemiology
- LV (or biventricular) dilation + impaired systolic function in absence of CAD, valvular disease, or pericardial disease
- LVEF <40% (or severely reduced)
- Prevalence: ~1 in 250 adults; most common cardiomyopathy in children (58% of pediatric cases; peak age 18 months)
- Males and females approximately equally affected, except X-linked forms (male predominance)
(Goldman-Cecil Medicine)
Etiology & Genetics
Genetic DCM (20-50% of cases):
- Autosomal dominant most common pattern
- TTN (titin) truncating mutations = single most common cause (~25% familial, ~18% sporadic)
- Also: MYH7, TNNT2, TPM1, ACTC1 (sarcomeric proteins - note: SAME genes cause HCM when gain-of-function; cause DCM when loss-of-function)
- Lamin A/C (LMNA): arrhythmogenic form - AF, conduction disease, VT/VF often PRECEDE heart failure; high SCD risk - ICD recommended even with LVEF >35%
- Filamin C (FLNC): mixed arrhythmogenic + dilated phenotype; lethal VAs in adolescents/young adults
- Dystrophin (DMD gene, X-linked): Duchenne and Becker muscular dystrophy
- Desmin: combined cardiac + skeletal myopathy; intermediate filament protein
High-risk genotypes for SCD (ICD warranted even before LVEF <35%):
DES, DSP, DSC2, DCG2, FLNC, LMNA, PKP2, PLN, RBM20, SCN5A, TMEM43
Acquired DCM causes:
| Cause | Key Detail |
|---|---|
| Viral myocarditis | Coxsackievirus B, enteroviruses, parvovirus B19, HHV-6; viral footprints persist to late-stage |
| Alcohol | 6 drinks/day for 5-10 years; acetaldehyde direct cardiotoxin; >50% improve with abstinence |
| Anthracyclines | Doxorubicin - overt HF in 5-10% at doses >450 mg/m²; dose-dependent; within first year |
| Trastuzumab | Up to 11% develop DCM; reversible with withdrawal |
| Peripartum | See below |
| Tachycardiomyopathy | Reverses once tachycardia controlled |
| Hemochromatosis, sarcoidosis, radiation, cocaine, nutritional deficiencies |
Clinical Presentation
- Symptoms of biventricular heart failure: exertional dyspnea, orthopnea, PND, peripheral edema
- First presentation may be sudden death or thromboembolic event (LV thrombus, especially at apex)
- Family history is important (30-40% familial clustering)
- May also present with arrhythmias (AF, VT, conduction disease) - especially with LMNA, FLNC mutations
Pathology
- Gross: Four-chamber dilation and hypertrophy; flabby, poorly contractile walls; mural thrombus (LV apex). Heart weight often >800 g (normal upper limit ~360 g)
- Histology: Nonspecific - myocyte hypertrophy + interstitial fibrosis (Masson trichrome shows blue collagen). No diagnostic features at end-stage
DCM gross specimen: massive biventricular dilation; apparent wall thinning despite overall hypertrophy (>800 g). Harrison's 22E, Fig. 267-6
Diagnosis
- TTE (transthoracic echo): first line; LV dilation, reduced LVEF, wall motion abnormalities
- CMR: gold standard for tissue characterization; mid-wall late gadolinium enhancement (LGE) is a key prognostic marker and predicts VT; 2023 ESC endorses CMR as primary diagnostic modality
- Biomarkers: BNP/NT-proBNP elevated; high-sensitivity troponin (consensus across all guidelines)
- ECG: LBBB, conduction defects, arrhythmias; LBBB + DCM = indication for CRT evaluation
- Genetic testing: Recommended when diagnostic phenotype established; essential for cascade family screening
Management: DCM
Heart Failure with Reduced EF (HFrEF) - Guideline-Directed Medical Therapy (GDMT):
All four pillars of the 2022 AHA/ACC/HFSA HF guideline apply:
| Drug Class | Example | Key Points |
|---|---|---|
| ACE inhibitor / ARB / ARNI | Sacubitril/valsartan (preferred over ACEi if tolerated) | Reduce mortality; sacubitril/valsartan superior to enalapril (PARADIGM-HF) |
| Beta-blocker | Carvedilol, bisoprolol, metoprolol succinate | Reduce SCD; carvedilol start 6.25 mg BID, titrate to 25 mg BID |
| MRA | Spironolactone, eplerenone | Reduce mortality and hospitalizations |
| SGLT2 inhibitor | Empagliflozin, dapagliflozin | Reduce HF hospitalizations/CV death (HFrEF + HFpEF); now standard of care |
2023 ESC novel recommendation: HF therapies may be initiated in asymptomatic patients with subclinical DCM/NDLVC phenotype to prevent adverse ventricular remodeling (Class IIb C).
Device therapy:
- CRT (cardiac resynchronization therapy): LVEF ≤35% + LBBB + QRS ≥150 ms (Class I) or QRS 120-149 ms (Class IIa)
- ICD for primary prevention: Standard threshold LVEF ≤35% despite 3+ months of GDMT
- Genotype-driven ICD (earlier implantation): LMNA or FLNC variants - ICD before LVEF <35% if ≥2 risk factors (male sex, NSVT, >10% fibrosis on CMR, non-missense mutations). 2023 ESC recommends SCD risk stratification by genotype even when LVEF >35% (Class IIa B)
Anticoagulation: In presence of LV thrombus, AF, or severe ventricular dilation
(Goldman-Cecil Medicine; Harrison's 22E; 2025 systematic review [PMID: 39674807])
PART 3: HYPERTROPHIC CARDIOMYOPATHY (HCM)
Definition & Epidemiology
- Unexplained LV hypertrophy in absence of abnormal loading conditions (valve disease, HTN, congenital defects)
- Prevalence: 0.2-0.5% worldwide; all racial groups
- Most common inherited cardiac disease
Genetics & Pathogenesis
- Autosomal dominant, gain-of-function mutations in sarcomeric contractile proteins
-
1,400 mutations in >9 genes identified; 50-60% of cases have identifiable sarcomere variants
- The Big 3 genes (account for 70-80%):
- MYH7 (beta-myosin heavy chain) - most common
- MYBPC3 (myosin-binding protein C)
- TNNT2 (troponin T)
- Mechanism: hypercontractility → increased energy use → net negative energy balance → compensatory hypertrophy + fibrosis
- Same genes (MYH7, etc.) cause DCM when loss-of-function; cause HCM when gain-of-function
Pathology
Gross:
- Massive hypertrophy WITHOUT ventricular dilation
- 90%: Asymmetric septal hypertrophy - septum disproportionately thicker than LV free wall
- 10%: concentric hypertrophy
- LV cavity: "banana-shaped" on longitudinal section
- Fibrous endocardial plaque in LV outflow tract (from SAM contact with septum)
- Systolic anterior motion (SAM) of the mitral valve → LV outflow tract obstruction (LVOTO) in 1/3 of cases
Histology (hallmark triad):
- Extreme myocyte hypertrophy
- Myocyte disarray (haphazard, chaotic architecture) - the pathognomonic feature
- Interstitial fibrosis
(A) Septal muscle compressing LV outflow tract into a "banana" shape; LA enlarged; fibrous endocardial plaque visible (arrow). (B) Myocyte disarray and interstitial fibrosis on H&E. Robbins & Kumar Basic Pathology, Fig. 9.27
Clinical Features
- Often asymptomatic or mild; presents during postpubertal growth spurt or young adulthood
- Triad of symptoms: exertional dyspnea, chest pain (angina without CAD), syncope
- Harsh systolic ejection murmur at LLSB; increases with Valsalva, standing (decreased preload); decreases with squatting, leg raise
- Sudden cardiac death - most important complication; HCM is responsible for ~1/3 of SCD in athletes under 35
ECG findings in HCM
- LVH pattern
- Deep narrow Q waves in lateral and inferior leads (septal depolarization)
- T-wave abnormalities
- AF is common (20-25%)
2024 AHA/ACC HCM Guidelines - Key Updates (PMID: 38718139)
Diagnosis:
- TTE: first-line; LV wall thickness ≥15 mm (or ≥13 mm with family history/genetic variant)
- CMR: when TTE inconclusive; superior tissue characterization; LGE ≥15% of LV mass = major SCD risk factor
Medical management of obstructive HCM (oHCM):
| Step | Drug | Mechanism |
|---|---|---|
| 1st line | Beta-blockers (metoprolol, atenolol) | Reduce HR, prolong diastolic filling, reduce LVOT gradient |
| 2nd line | Verapamil or diltiazem | Improve diastolic relaxation |
| 3rd line | Disopyramide (+ beta-blocker) | Negative inotrope |
| NEW (2024) | Mavacamten (Camzyos) | Cardiac myosin inhibitor; reduces hypercontractility and LVOT gradient; first-in-class disease-modifying therapy |
Mavacamten (major 2024 guideline addition):
- Selective, reversible cardiac myosin ATPase inhibitor
- Approved by FDA; EXPLORER-HCM trial showed significant improvement in LVOT gradient, symptoms, and exercise capacity
- Monitoring: Echo every 4 weeks for first 12 weeks (FDA requirement); discontinue if LVEF drops <50%
- Contraindicated with strong CYP3A4 inhibitors; teratogenic
Septal reduction therapy (for refractory oHCM):
- Surgical septal myectomy (Morrow procedure) - gold standard; preferred for younger patients
- Alcohol septal ablation (ASA): catheter-based; intracoronary ethanol injection causes controlled septal MI; preferred for older/high surgical risk patients
- Indication: LVOT gradient ≥50 mmHg at rest or with provocation + refractory symptoms (NYHA III-IV) despite maximal medical therapy
SCD prevention / ICD indications (2024 ACC/AHA):
Primary prevention ICD - Class IIa if ≥1 major risk factor:
- Family history of SCD at young age (≤40 years)
- Unexplained syncope (especially within 6 months)
- LVEF <50%
- Apical aneurysm with transmural scar
- Extensive LGE on CMR (≥15% of LV mass)
- NSVT on ambulatory monitoring
Use the HCM Risk-SCD calculator to estimate 5-year SCD risk - includes LVOT gradient and LA diameter (2024 addition).
AF management in HCM (2024 guidelines):
- Anticoagulation recommended in ALL HCM patients with AF regardless of CHA2DS2-VASc score
- DOACs (direct oral anticoagulants) = first-line
- Vitamin K antagonists = second-line
- Rate control: beta-blockers, verapamil, diltiazem
- Rhythm control: amiodarone (effective in HCM); catheter ablation (higher relapse rate vs non-HCM)
Physical activity (2024 guidelines - paradigm shift):
- HCM patients should engage in mild to moderate recreational exercise for cardiovascular health
- Competitive/vigorous sports: individualized shared decision-making with HCM expert (no longer blanket prohibition)
- 2025 ACC/AHA Sports Participation Guidelines: athletes with confirmed HCM - "Reasonable to consider participation" (vs 2015 guideline: "Should not participate")
Family screening (2024 ACC/AHA):
- First-degree relatives of HCM proband: ECG + TTE every 12-18 months during adolescence; every 3-5 years in adults
- Genetic testing: recommended when clinical diagnosis established; valuable for cascade screening
- Genotype positive/phenotype negative: annual clinical evaluation
PART 4: RESTRICTIVE CARDIOMYOPATHY (RCM)
Definition
Decreased ventricular compliance → impaired diastolic filling. Systolic function usually preserved. Least common primary cardiomyopathy.
Pathology
- Ventricles normal or slightly enlarged (NOT dilated)
- Both atria markedly dilated (consequence of chronic elevated filling pressures)
- Myocardium is firm
- Mimics constrictive pericarditis clinically (must distinguish - important exam distinction!)
| Feature | Restrictive CMP | Constrictive Pericarditis |
|---|---|---|
| Pericardium | Normal | Thickened/calcified |
| Kussmaul sign | Present | Present |
| Pericardial knock | Absent | Present |
| Systemic JVD equalization | Less common | Characteristic |
| Treatment | Cause-specific | Pericardiectomy |
Causes
1. Cardiac amyloidosis (most clinically important)
| Type | Protein | Key Features |
|---|---|---|
| AL amyloid (primary) | Immunoglobulin light chains | Plasma cell dyscrasia (MM); light chains also directly cardiotoxic to myocytes |
| ATTR-wild type (senile systemic amyloidosis) | Normal transthyretin (TTR) | Older men (>60); carpal tunnel syndrome, spinal stenosis, biceps tendon rupture - classic clues |
| ATTR-hereditary | Mutant TTR | 4% of African Americans carry the Val122Ile mutation → 4-fold increased risk; younger onset |
Clues to cardiac amyloidosis on exam:
- "Sparkling" myocardium on echo (granular appearance)
- Low-voltage ECG despite LV hypertrophy on echo (pseudo-LVH)
- Bilateral carpal tunnel syndrome + HF
- Spinal stenosis + HF
- Macroglossia (AL type)
- "Cherry blossoms" (HCC-like) on CMR with specific LGE pattern
Diagnosis of ATTR:
- Pyrophosphate (PYP/DPD) bone scintigraphy - can diagnose ATTR noninvasively (Perugini Grade 2-3) if monoclonal protein screen is negative (SPEP, UPEP, serum FLC)
- If monoclonal protein present → biopsy needed to exclude AL
Treatment of ATTR cardiomyopathy (2025 ACC Concise Clinical Guidance, PMID: 41171219):
- Tafamidis (Vyndaqel) - TTR stabilizer; FDA-approved; reduces mortality (ATTR-ACT trial); first-line
- Acoramidis - TTR stabilizer; newer, approved 2023-2024
- Vutrisiran (siRNA) - TTR silencer; reduces TTR production; alternative approach
- SGLT2 inhibitors and MRAs: now endorsed as broadly effective HF therapies in ATTR
- Standard HF therapy: diuretics (symptom relief); digoxin relatively contraindicated (binds amyloid fibrils); calcium channel blockers generally avoided
2. Endomyocardial fibrosis
- Most common RCM worldwide (children/young adults, sub-Saharan Africa, tropical regions)
- Diffuse fibrosis of ventricular endocardium and subendocardium
- Often involves tricuspid and mitral valves
- Linked to nutritional deficiencies and helminthic infections
3. Loeffler endomyocarditis
- No geographic predilection
- Peripheral hypereosinophilia + eosinophilic tissue infiltrates
- Eosinophil major basic protein → endocardial necrosis → mural thrombus → organization → fibrosis
- Stages: necrotic → thrombotic → fibrotic
4. Other causes: Radiation fibrosis, sarcoidosis, storage diseases (mucopolysaccharidoses, Gaucher, Fabry), hemochromatosis, idiopathic
PART 5: ARRHYTHMOGENIC CARDIOMYOPATHY (ACM/ARVC)
Definition
Genetically determined structural cardiomyopathy with prominent arrhythmic presentation; fibro-fatty replacement of myocardium - predominantly RV but may involve LV (ALVC) or be biventricular.
- Responsible for up to 22% of SCD in athletes
- Prevalence: ~1 in 5,000
Genetics
- Desmosomal protein mutations (cell-cell adhesion):
- PKP2 (plakophilin-2) - most common
- DSP (desmoplakin)
- DSG2 (desmoglein-2)
- DSC2 (desmocollin-2)
- JUP (junction plakoglobin)
- Non-desmosomal: LMNA, desmin, others
- Naxos disease: autosomal recessive; PKP2 mutation; clinical triad = ARVC + woolly hair + palmoplantar keratoses (first described on Greek island of Naxos)
- Carvajal syndrome: DSP mutation; ARVC + woolly hair + palmoplantar keratosis + DILATED (not just RV) cardiomyopathy
Pathology
- RV markedly dilated; fibro-fatty replacement of RV free wall (almost transmural)
- LV may be involved to lesser extent
- "Triangle of dysplasia": RV inflow, outflow, and apex
(A) RV dilated with fibro-fatty replacement of free wall. (B) Masson trichrome: RV myocardium (red) replaced by fibrosis (blue) and fat (arrow). Robbins & Kumar Basic Pathology, Fig. 9.26
Diagnosis: Task Force Criteria (Major and Minor)
Diagnosis requires 2 major, 1 major + 2 minor, or 4 minor criteria across 6 categories:
| Category | Major | Minor |
|---|---|---|
| Structural/functional (RV) | RVEF ≤40% or RV dilation on echo/CMR/angiography | RVEF 41-45% or milder dilation |
| Tissue characterization | Fibro-fatty replacement on biopsy | |
| Repolarization | T-wave inversion V1-V3 (age >14, no RBBB) | T-wave inversion V1-V2 |
| Depolarization | Epsilon wave (V1-V3); terminal QRS >55 ms in V1-V3 | Late potentials on signal-averaged ECG |
| Arrhythmia | Sustained or NSVT with LBBB morphology, superior axis | LBBB-type VT from RVOT; >500 PVCs/24 hrs |
| Family history | ARVC in first-degree relative; pathogenic desmosomal mutation |
Key ECG features:
- Epsilon wave (classic) - small high-frequency deflection after QRS in V1-V3
- T-wave inversions V1-V3 (most sensitive)
- Terminal QRS prolongation >55 ms in V1-V3 (distinct from RBBB)
- VT with LBBB morphology + superior or inferior axis = arises from RV
Management
- Lifestyle: Avoid strenuous exercise/competitive sports (especially endurance); exercise accelerates disease progression by increasing RV wall stress
- Beta-blockers: first-line for arrhythmia prevention
- Antiarrhythmics: sotalol (160-240 mg/day), amiodarone (200 mg/day maintenance); flecainide (endorsed by 2023 ESC for atrial and ventricular arrhythmias in ARVC)
- ICD indications:
- Definite: prior SCA, sustained VT, LVEF ≤35%
- Primary prevention (Class IIa): LVEF ≤50% + ≥1 major risk factor (NSVT, inducible VT, RV dysfunction, male sex, proband, >1 desmosomal mutation)
- Catheter ablation: for drug-refractory VT or frequent ICD shocks
- Heart failure therapy (ACEi, beta-blocker, diuretics) if progresses to severe HF
- Anticoagulation: if AF, marked ventricular dilation, or ventricular aneurysms
- Cardiac transplantation: for refractory end-stage disease
2025 ACC/AHA Sports Participation update:
- PKP2 ACM: risks may outweigh benefits (sports caution)
- Non-PKP2 ACM: can consider participation with shared decision-making
- Genotype positive/phenotype negative: reasonable to consider participation
(Goldman-Cecil Medicine; Fuster and Hurst's The Heart 15E; Harrison's 22E)
PART 6: PERIPARTUM CARDIOMYOPATHY (PPCM)
Definition & Diagnostic Criteria
DCM occurring in the last trimester of pregnancy OR within months of delivery, in the absence of prior heart disease, with:
- LVEF <45%
- No other identifiable cause of HF
Epidemiology
- US: 1 per ~3,000 live births; Haiti/Africa: 1 per ~300 live births (much higher in Black women)
- Risk factors: older maternal age, hypertension, preeclampsia, African-American race, multifetal pregnancies
Pathogenesis (multifactorial)
- Genetic: 15% have rare truncating variants (2/3 involve TTN); similar rate to idiopathic DCM → PPCM may be an unmasked familial DCM
- Prolactin cleavage: Oxidative stress → prolactin cleaved into a pro-inflammatory, vasotoxic 16-kDa fragment → endothelial and cardiomyocyte dysfunction
- Anti-angiogenic: Placenta releases soluble fms-like tyrosine kinase-1 (sFlt-1) → angiogenic imbalance
- Overlap with preeclampsia (~25% of PPCM vs 5% population)
Prognosis
- ~40-70% have normalization of LVEF within 5 years (better than idiopathic DCM)
- Patients with persistent LV dysfunction at baseline or African-American race: worse outcomes
- Subsequent pregnancy: Risk of recurrence in all patients, even those who normalize LV function
- Normalized LV function: HF recurs in 20% of subsequent pregnancies (mortality 0% in this group)
- Persistent LV dysfunction: HF recurs in 40%; maternal mortality 19%
Treatment
| Drug | Comment |
|---|---|
| Hydralazine + isosorbide dinitrate | Vasodilators safe in pregnancy (ACEi/ARBs/ARNI contraindicated - teratogenic) |
| Beta-blockers | Safe in pregnancy (carvedilol, metoprolol) |
| Diuretics | Judicious use for symptom relief |
| Bromocriptine (dopamine agonist) | Inhibits prolactin; small studies show improvement in LVEF; still controversial |
| Anticoagulation | If severe LV dysfunction; LMWH preferred (not warfarin - teratogenic in first trimester) |
| Digoxin | Safe in pregnancy; used for rate control |
(Creasy & Resnik's Maternal-Fetal Medicine; Goldman-Cecil Medicine)
PART 7: STRESS (TAKOTSUBO) CARDIOMYOPATHY
Overview
- Triggered by acute severe emotional, psychological, or physical stress
- Characterized by transient apical LV ballooning and dysfunction that is usually reversible
- "Takotsubo" = Japanese octopus trap (the shape of the LV during systole)
Epidemiology
- Predominantly postmenopausal women (>90% of cases)
- Accounts for 2-3% of all suspected ACS presentations
- COVID-19 increased incidence: 7.8% of ACS presentations during pandemic vs 1.5-1.8% pre-pandemic
Key Features (how to distinguish from STEMI)
| Feature | Takotsubo | STEMI |
|---|---|---|
| Troponin elevation | Mild (low) | Marked |
| ST elevation | May be present, diffuse | Focal, territory-matching |
| Coronary angiography | Normal (no culprit lesion) | Culprit stenosis/occlusion |
| Wall motion abnormality | Extends beyond single coronary territory (apical ballooning) | Territory of one coronary artery |
| Resolution | Complete in days-weeks | Permanent (infarct) |
| LVEF | Acutely reduced (~30%), recovers | Depends on infarct size |
Management
- Supportive care: beta-blockers, ACEi, diuretics for HF
- Avoid catecholamines (worsens condition)
- If cardiogenic shock: IABP or Impella; avoid vasopressors if possible
- Anticoagulation if LV thrombus suspected
- Prognosis: generally excellent with full recovery; mortality ~4-5% in hospitalized patients (usually comorbid conditions)
PART 8: OTHER / SECONDARY CARDIOMYOPATHIES
Infiltrative/Storage Diseases Mimicking HCM ("Pseudo-hypertrophy")
| Disease | Key Feature | Treatment |
|---|---|---|
| Fabry disease | X-linked; alpha-galactosidase A deficiency; glycolipid accumulation; classic: angiokeratomas, acroparesthesias, corneal opacities | Enzyme replacement therapy (agalsidase); oral chaperone migalastat |
| Pompe disease | Glycogen storage (acid maltase deficiency); massive hypertrophy in infants; short PR interval | Enzyme replacement |
| Anderson-Fabry | Above; also causes HCM phenotype | |
| Danon disease | LAMP2 mutation; X-linked; HCM + Wolff-Parkinson-White + skeletal myopathy | Supportive; transplant |
| LEOPARD/Noonan syndrome | RAS/MAPK pathway mutations; HCM phenotype + lentigines, ECG abnormalities, hypertelorism | Supportive |
| Cardiac sarcoidosis | Granulomatous infiltration; heart block, VT, DCM phenotype; MRI shows patchy LGE | Steroids; ICD |
Chemotherapy-Induced Cardiomyopathy (Cardio-oncology)
| Agent | Mechanism | Management |
|---|---|---|
| Anthracyclines (doxorubicin) | Oxidative damage, topoisomerase II inhibition; dose-dependent; overt HF at >450 mg/m² | Dexrazoxane (cardioprotective); carvedilol or enalapril prophylaxis |
| Trastuzumab | HER2 pathway; up to 11% DCM; reversible with withdrawal | Withdraw + standard HFrEF therapy |
| Immune checkpoint inhibitors | T-cell off-target myocarditis; rare but potentially fatal | High-dose steroids; stop immunotherapy |
| Tyrosine kinase inhibitors (sunitinib) | Reduce LVEF; more with CAD | Withdraw; standard HFrEF therapy |
PART 9: MYOCARDITIS (Related Entity)
Overview
Myocardial damage due to inflammatory infiltrates. Important because:
- Can cause acute HF
- Can progress to DCM (DCM is considered a potential sequela of viral myocarditis)
Causes
| Category | Examples |
|---|---|
| Viral (most common in US) | Coxsackievirus A and B, enteroviruses; parvovirus B19, HHV-6; CMV, HIV, influenza |
| Bacterial | Diphtheria (Corynebacterium diphtheriae), Lyme disease (Borrelia) |
| Parasitic | Chagas disease (Trypanosoma cruzi) - most important worldwide cause of cardiomyopathy in Latin America |
| Autoimmune | SLE, polymyositis; giant cell myocarditis (rare but fatal) |
| Toxic | Anthracyclines, cocaine |
| Eosinophilic | Drug hypersensitivity, hypereosinophilic syndrome |
Giant Cell Myocarditis
- Rapidly fatal without treatment
- Diagnosis: endomyocardial biopsy (multinucleated giant cells)
- Treatment: prolonged immunosuppression (cyclosporine + steroids); transplant if refractory
Chagas Cardiomyopathy
- Trypanosoma cruzi - vector: Reduviid (kissing) bug
- Affects 8-10 million in Latin America; leading cause of cardiomyopathy in endemic regions
- Chronic phase: dilated cardiomyopathy + right bundle branch block + left anterior fascicular block (classic ECG pattern) + apical aneurysm + thromboembolic events
- Treatment: Benznidazole/nifurtimox (antiparasitic); standard HFrEF therapy; ICD for VT/VF
PART 10: SUMMARY COMPARISON TABLE (HIGH-YIELD EXAM REFERENCE)
| Feature | DCM | HCM | RCM | ARVC |
|---|---|---|---|---|
| LV size | Dilated (4-chamber) | Normal-enlarged (thick-walled) | Normal/small | RV dilated |
| Wall thickness | Thin (relative) | Massively thick | Variable | RV thin (fibro-fatty) |
| Systolic function | Severely impaired (LVEF <40%) | Preserved (LVEF 50-80%) | Preserved | RV impaired |
| Diastolic function | Secondarily impaired | Primarily impaired (stiff) | Severely impaired | - |
| Pathognomonic finding | 4-chamber dilation | Myocyte disarray + septal hypertrophy | Biatrial dilation + firm myocardium | Fibro-fatty RV replacement |
| Key histology | Myocyte hypertrophy + fibrosis | Disarray + fibrosis | Amyloid/fibrosis | Fibroadipose tissue |
| Murmur | Functional MR | Harsh systolic ejection murmur (LLSB); increases with Valsalva | None/S3-S4 | None |
| Top genetic causes | TTN, LMNA, DMD (loss-of-function) | MYH7, MYBPC3, TNNT2 (gain-of-function) | TTR, J chain | PKP2, DSP, DSG2 (desmosomal) |
| Key drug (new) | SGLT2i, sacubitril/valsartan | Mavacamten | Tafamidis/acoramidis (ATTR) | Beta-blocker + sotalol |
| SCD risk | Moderate (LMNA/FLNC high) | High (young athletes) | Low-moderate | Very high (athletes) |
| ICD threshold | LVEF ≤35% (earlier for LMNA/FLNC) | ≥1 major risk factor | Cause-specific | Per Task Force criteria |
| Sports restriction | Shared decision-making (2025) | Shared decision-making (2024) | Low-moderate activity | Avoid strenuous exercise |
PART 11: GUIDELINE SUMMARY REFERENCE
| Guideline | Year | Key Highlights |
|---|---|---|
| 2023 ESC Guidelines on Cardiomyopathies | 2023 | New NDLVC entity; CMR as primary diagnostic modality; genotype-driven SCD risk stratification; HF therapy in asymptomatic subclinical phenotype (IIb C); flecainide endorsed in ARVC; anticoagulation in HCM/amyloid regardless of risk score |
| 2024 AHA/ACC HCM Guidelines | 2024 | Mavacamten introduced (cardiac myosin inhibitor); shared decision-making for sports; DOACs first-line in HCM+AF regardless of CHA2DS2-VASc; updated SCD risk factors (LGE ≥15%, apical aneurysm); HCM Risk-SCD calculator updated |
| 2025 ACC ATTR Guidance | 2025-2026 | Tafamidis and acoramidis (TTR stabilizers) + vutrisiran (siRNA silencer) now endorsed; SGLT2i and MRA for all ATTR-CM patients; PYP scintigraphy for noninvasive diagnosis |
| 2025 ACC/AHA Sports Participation | 2025 | Paradigm shift - cardiomyopathy no longer uniformly excludes athletes; gene+/phenotype- can participate; confirmed HCM = "reasonable to consider participation" |
| 2022 AHA/ACC/HFSA HF Guidelines | 2022 | Four pillars of GDMT (ACEi/ARB/ARNI + BB + MRA + SGLT2i) for ALL HFrEF including DCM |
| 2025 ESC Myocarditis Guidelines | 2025 | Updated diagnostic criteria; genetic screening for family members; new CMR Lake Louise criteria |
Sources: Goldman-Cecil Medicine; Harrison's Principles of Internal Medicine, 22E; Robbins & Kumar Basic Pathology (9th ed.); Robbins, Cotran & Kumar Pathologic Basis of Disease; Creasy & Resnik's Maternal-Fetal Medicine; Fuster & Hurst's The Heart, 15E; Tintinalli's Emergency Medicine; 2024 AHA/ACC HCM Guideline (PMID: 38718139); DCM Guidelines Systematic Review 2025 (PMID: 39674807); 2025 ACC ATTR Guidance (PMID: 41171219); 2023 ESC Cardiomyopathy Guidelines (Eur Heart J 44:3503-3626)
This is a shared conversation. Sign in to Orris to start your own chat.