An 18-year-old lady presented with low-grade fever for 14 days. She had a history of tooth extraction 20 days back and is a known case of valvular heart disease. a. What is your diagnosis? Discuss the pathophysiology. b. Clinical features. c. Investigations and management. d. Prevention and prophylaxis of the above disease.
"infective endocarditis"[MeSH Terms] AND "prophylaxis"[MeSH Terms]
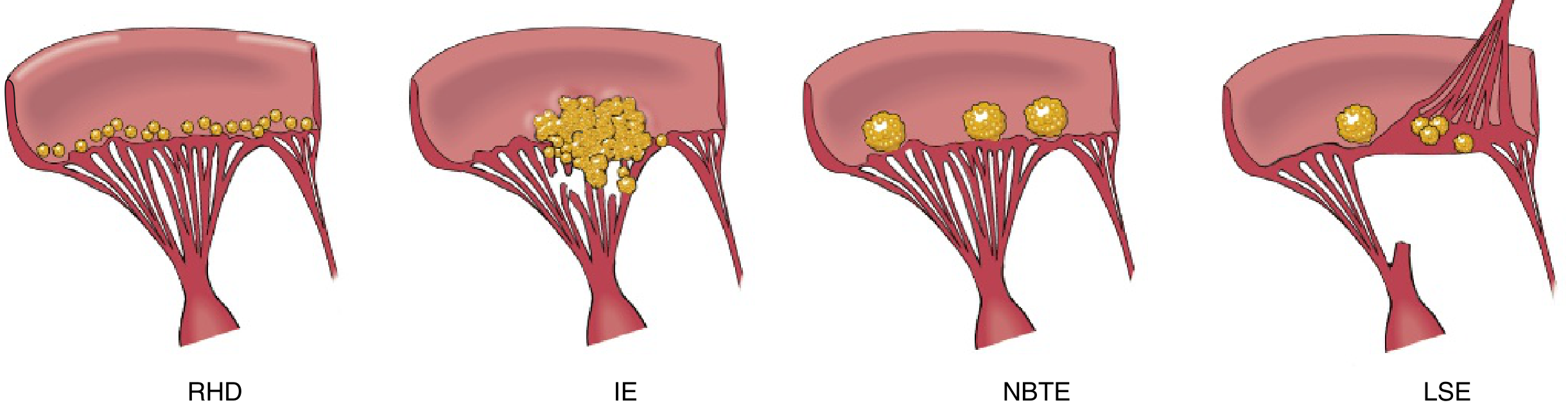
Infective Endocarditis (IE)
a. Diagnosis and Pathophysiology
Diagnosis
- Predisposing cardiac lesion (valvular heart disease)
- Bacteremia from a dental procedure (tooth extraction 20 days prior)
- Low-grade fever for 14 days (subacute course)
Pathophysiology
b. Clinical Features
Systemic Features
- Fever - most consistent finding (low-grade in subacute, high with chills in acute)
- Constitutional symptoms: fatigue, malaise, weight loss, anorexia
- Musculoskeletal complaints: myalgias, arthralgias, back pain (~50%)
- Splenomegaly (especially in subacute IE)
Cardiac Features
- New or changing heart murmur (present in ~90% of left-sided lesions)
- Signs of heart failure (dyspnea, orthopnea) if valve destruction is severe
- Conduction defects (PR prolongation) suggest aortic root/ring abscess
Peripheral Embolic and Immunological Manifestations (classic "peripheral stigmata")
| Feature | Description | Mechanism |
|---|---|---|
| Petechiae | On conjunctivae, palate, skin | Microemboli / vasculitis |
| Splinter hemorrhages | Subungual, linear, non-blanching | Microemboli in nail bed vessels |
| Roth spots | Boat-shaped retinal hemorrhages with pale center | Immune complex vasculitis of retinal vessels |
| Osler nodes | Painful, tender, raised nodules on fingertips/toes | Immune complex deposition (vasculitis) |
| Janeway lesions | Painless, flat, erythematous lesions on palms/soles | Septic emboli |
| Clubbing | Late sign in prolonged SBE | Chronic hypoxia/cytokine effect |
Renal Manifestations
- Microscopic hematuria and proteinuria (focal embolic nephritis or immune complex glomerulonephritis)
- Renal failure in severe cases
Neurological Complications
- Embolic stroke or TIA
- Cerebral abscess
- Mycotic aneurysm (at arterial bifurcations - from septic emboli invading arterial wall)
- Meningitis
c. Investigations and Management
Investigations
- Three sets from three different sites, drawn at least 1 hour apart, before starting antibiotics
- In most untreated patients, all cultures are positive
- In ~10% of cases, cultures are negative (prior antibiotics, fastidious organisms)
- Positive cultures fulfill a major Duke criterion
| Major Criteria | Minor Criteria |
|---|---|
| Positive blood cultures (typical organisms in ≥2 cultures, or persistently positive) | Predisposing heart condition or IV drug use |
| Evidence of endocardial involvement (echo: vegetation, abscess, new valve dehiscence; new valvular regurgitation) | Fever >38°C |
| Vascular phenomena (emboli, Janeway lesions, mycotic aneurysm, conjunctival hemorrhage) | |
| Immunologic phenomena (Osler nodes, Roth spots, RF positive) | |
| Microbiological evidence not meeting major criterion |
- Definite IE: 2 major, OR 1 major + 3 minor, OR 5 minor criteria
- Possible IE: 1 major + 1 minor, OR 3 minor
- Rejected: Firm alternative diagnosis, or resolution with ≤4 days antibiotics
- Transthoracic echo (TTE): First line; detects vegetations, valve regurgitation, abscess
- Transesophageal echo (TEE): Superior sensitivity (>90%); mandatory if TTE negative but suspicion high, in prosthetic valves, or when periannular complications suspected
- CBC: anemia (chronic disease), leukocytosis (more in acute)
- ESR, CRP raised
- Urinalysis: hematuria, proteinuria
- Rheumatoid factor: positive in subacute IE (minor Duke criterion)
- Renal function tests: BUN, creatinine
- LFTs
- Chest X-ray: Evidence of heart failure (cardiomegaly, pulmonary edema), or septic emboli in right-sided IE
- ECG: PR prolongation, AV block (suggests perivalvular abscess)
- CT/MRI brain: For neurological complications
- CT abdomen: Splenic/renal infarcts, mycotic aneurysms
Management
| Organism | First-line Treatment |
|---|---|
| Streptococcus viridans (penicillin-sensitive) | Penicillin G 12-18 million U/day IV for 4 weeks; OR Amoxicillin/ampicillin for 4 weeks (± gentamicin for first 2 weeks for synergy) |
| Streptococcus viridans (penicillin-resistant) | Penicillin G + gentamicin for 4-6 weeks |
| S. aureus (MSSA) | Nafcillin/flucloxacillin for 4-6 weeks |
| S. aureus (MRSA) | Vancomycin for 6 weeks |
| Enterococci | Ampicillin + gentamicin for 4-6 weeks |
| Culture-negative | Empirical: Ampicillin + gentamicin OR vancomycin + gentamicin |
- Penicillin-allergic patients: ceftriaxone or vancomycin as alternatives (clindamycin is no longer recommended as an alternative due to C. difficile risk per 2021 AHA update)
- Daily cardiac exam for new murmurs, widening pulse pressure
- Serial ECGs for conduction abnormalities
- Repeat blood cultures (especially in S. aureus IE)
- Repeat TTE/TEE if: persistent fever, new murmur, embolic events, signs of heart failure
- Monitor renal function (especially if aminoglycosides used); gentamicin trough should be <1-3 µg/mL
- Heart failure due to severe valve regurgitation (most common surgical indication)
- Periannular abscess or fistula
- Persistent bacteremia despite appropriate antibiotics (>5-7 days)
- Fungal endocarditis
- Large vegetations (>10 mm) with high embolic risk
- Prosthetic valve endocarditis with instability
- Treat heart failure (diuretics, afterload reduction)
- Monitor neurological status
- Addiction medicine consultation if injection drug use is involved
- Avoid anticoagulation (increases hemorrhagic transformation risk in embolic strokes)
d. Prevention and Prophylaxis
AHA 2021 Guidelines - Antibiotic Prophylaxis
- Prosthetic cardiac valves (including transcatheter-implanted) or prosthetic material used for valve repair
- Previous infective endocarditis
- Unrepaired cyanotic congenital heart disease or palliated with shunts/conduits
- Repaired congenital heart disease with residual defects at or adjacent to prosthetic material
- Cardiac transplant recipients with valve regurgitation
- Left ventricular assist devices and implantable hearts (added in 2021)
Note: Valvular heart disease (rheumatic, degenerative) alone is NOT currently listed in the AHA highest-risk category requiring prophylaxis - however, many centers and guidelines (including ESC) still recommend prophylaxis for high-risk valvular lesions. In developing countries with endemic rheumatic heart disease, this patient would commonly receive prophylaxis.
Prophylaxis Regimens
| Situation | Antibiotic | Dose (Adult) | Timing |
|---|---|---|---|
| Able to take oral medications | Amoxicillin (first choice) | 2 g PO | 30-60 min before procedure |
| Penicillin allergy (non-anaphylactic) | Cephalexin | 2 g PO | 30-60 min before |
| Penicillin allergy (anaphylactic) | Azithromycin or clarithromycin | 500 mg PO | 30-60 min before |
| Unable to take oral | Ampicillin or cefazolin/ceftriaxone | 2 g IV/IM | 30-60 min before |
| Penicillin allergy + unable to take oral | Cefazolin or ceftriaxone | 1 g IV/IM | 30-60 min before |
Clindamycin has been removed as a recommended alternative (2021 AHA update) due to Clostridioides difficile risk. Doxycycline was added as an alternative in truly penicillin-allergic patients.
General Prevention Strategies
- Maintain excellent oral hygiene - regular dental care reduces routine bacteremia from daily activities (brushing, chewing); this may reduce IE risk more than procedural prophylaxis alone
- Aseptic technique during all invasive procedures
- Avoid unnecessary IV access and catheters
- Treat remote infections promptly (dental caries, skin infections, UTIs) before they cause bacteremia
- Patient education: Patients with high-risk cardiac lesions should inform all healthcare providers of their cardiac status before any invasive procedure
- Rheumatic fever prophylaxis (benzathine penicillin G monthly IM injections) prevents recurrent rheumatic heart disease and thereby reduces the pool of patients at risk for IE
- Harm reduction for injection drug users: needle exchange programs, addiction treatment
- Robbins & Kumar Basic Pathology, p. 368 (Pathogenesis & Morphology)
- Braunwald's Heart Disease, 11th ed., pp. 826+ (Clinical Presentation, Prevention, Duke Criteria)
- Goldman-Cecil Medicine, pp. 700-701+ (Pathobiology, Clinical Manifestations, Diagnosis, Management)
A 72-year-old diabetic patient presents with fever (100.8°F) for 5 days, marked hyperglycemia (680 mg/dL), fruity breath odor, Kussmaul breathing, serum Na⁺ 120 mEq/L, K⁺ 6.8 mEq/L, arterial blood pH 7.15. a. What is your diagnosis? Discuss the pathophysiology. b. Risk factors and clinical features. c. Management. d. What advice will you give to avoid recurrence?
"diabetic ketoacidosis"[MeSH Terms] AND "management"[MeSH Terms]
Diabetic Ketoacidosis (DKA)
a. Diagnosis and Pathophysiology
Diagnosis
- Hyperglycemia: Glucose 680 mg/dL (>350 mg/dL is DKA threshold)
- Ketosis: Fruity (acetone) breath odor, ketonemia/ketonuria ≥3.0 mmol/L
- Metabolic acidosis: Arterial pH 7.15 (<7.30 confirms DKA; <7.1 = severe DKA)
DKA vs. HHS distinction: This patient has pH 7.15, significant ketones, and glucose 680 mg/dL - fitting DKA. HHS typically has glucose >700 mg/dL, pH >7.30, minimal ketones, and much higher osmolality. Some Type 2 diabetics (like this patient) can present with a mixed DKA-HHS picture.
Pathophysiology
- Glucagon excess activates hepatic gluconeogenesis (using amino acids, lactate, glycerol as substrates)
- Glycogenolysis (liver) releases stored glucose
- Peripheral glucose uptake is blocked (no insulin to activate GLUT-4)
- Proteolysis in muscle accelerates → amino acids flood to liver as gluconeogenic substrates
- Result: Severe hyperglycemia (680 mg/dL)
- Insulin normally inhibits hormone-sensitive lipase in adipose tissue
- Without insulin → lipase activated → massive release of free fatty acids (FFAs) into circulation
- Long-chain FFAs are transported to the liver
- In normal metabolism, FFAs enter the TCA cycle via acetyl-CoA
- In DKA: malonyl-CoA (which normally inhibits mitochondrial FFA transport) is depleted because glucagon inhibits its synthesis → FFAs flood the mitochondria via carnitine acyltransferase
- Excess acetyl-CoA in liver exceeds TCA cycle capacity → diverted to ketogenesis: acetoacetate + β-hydroxybutyrate + acetone
- Peripheral tissues cannot use ketones fast enough → ketone accumulation
- Acetone = volatile → exhaled as fruity breath
- Hyperglycemia exceeds the renal threshold for glucose reabsorption (~180 mg/dL)
- Glycosuria → osmotic diuresis → profound loss of water, Na⁺, K⁺, Cl⁻, Mg²⁺, phosphate
- Average fluid deficit in severe DKA: 70-90 mL/kg body weight
- Average electrolyte deficits: Na⁺ 8-10 mEq/kg, K⁺ 5-7 mEq/kg
| Finding | Value | Explanation |
|---|---|---|
| Na⁺ 120 mEq/L | Low (pseudohyponatremia) | Hyperglycemia (680 mg/dL) shifts intracellular water into the extravascular space, diluting serum Na⁺. Correct: add 1.6 mEq/L Na⁺ for every 100 mg/dL glucose above 100 → true Na⁺ ≈ 120 + (5.8 × 1.6) ≈ 129 mEq/L (still low, reflecting total body Na⁺ depletion from osmotic diuresis) |
| K⁺ 6.8 mEq/L | Spuriously elevated | Metabolic acidosis drives H⁺ intracellularly → K⁺ exits cells in exchange → elevated serum K⁺. Despite this, total body K⁺ is severely depleted (osmotic diuresis loss). K⁺ will drop dramatically once insulin is given and acidosis corrects. |
b. Risk Factors and Clinical Features
Risk Factors / Precipitants
- Infections (most common - especially pneumonia, UTI, soft tissue infections)
- Inadequate insulin therapy / missed insulin doses / non-adherence
- New-onset Type 1 diabetes (presenting as DKA in ~25% of cases)
- Acute coronary syndrome or myocardial infarction
- Cerebrovascular accident, acute pancreatitis, pulmonary embolism
- Alcohol intoxication
- Endocrinopathies: Cushing syndrome, thyrotoxicosis, acromegaly (counter-regulatory excess)
- Severe burns, hyperthermia
- Drugs: Corticosteroids, SGLT-2 inhibitors (euglycemic DKA), clozapine, olanzapine, cocaine, thiazide diuretics, sympathomimetics
- Type 2 diabetes under physiological stress (sepsis, surgery, GI bleeding)
Patient Risk Factors in This Case:
- Type 2 diabetes (known diabetic, age 72)
- Active infection (fever for 5 days)
- Elderly age → impaired thirst perception and fluid intake
Clinical Features
- Polyuria, polydipsia, polyphagia (osmotic symptoms preceding the crisis)
- Weakness, lethargy, malaise
- Nausea, vomiting (very common)
- Abdominal pain (in ~50% - can mimic acute abdomen; in adults, often signifies a real abdominal precipitant)
- Blurred vision
- Weight loss
| Sign | Mechanism |
|---|---|
| Fever | Underlying infection (fever itself is NOT caused by DKA) |
| Kussmaul breathing (deep, rapid, sighing respiration) | Respiratory compensation for metabolic acidosis |
| Fruity/acetone breath odor | Exhaled acetone (volatile ketone) |
| Tachycardia | Dehydration, hypovolemia |
| Hypotension / orthostatic BP changes | Profound dehydration (3-5 L fluid deficit) |
| Dry skin and mucous membranes | Dehydration |
| Reduced skin turgor | Dehydration |
| Sunken eyes | Dehydration |
| Depressed sensorium / confusion / coma | Hyperosmolality, acidosis - correlates with severity |
| Reduced jugular venous pressure | Hypovolemia |
| Parameter | Mild | Moderate | Severe |
|---|---|---|---|
| Glucose (mg/dL) | >250 | >250 | >250 |
| Arterial pH | 7.25-7.30 | 7.00-7.24 | <7.00 |
| Serum HCO₃ (mEq/L) | 15-18 | 10-15 | <10 |
| Ketones (urine/serum) | Positive | Positive | Positive |
| Mental status | Alert | Alert/drowsy | Stupor/coma |
c. Management
1. Immediate Assessment and Monitoring
- Secure IV access (two large-bore IVs), cardiac monitor, pulse oximetry
- Urinary catheter (monitor urine output hourly)
- Nasogastric tube if vomiting/obtunded
- ICU/HDU admission for severe DKA (pH <7.1, altered consciousness, K⁺ abnormalities)
- Baseline labs: ABG, glucose, serum electrolytes (Na⁺, K⁺, Cl⁻, HCO₃⁻, BUN, creatinine, phosphate, Mg²⁺), CBC, urinalysis with ketones, blood cultures, CXR, ECG
- Calculate anion gap: AG = Na⁺ - (Cl⁻ + HCO₃⁻); normal 8-12 mEq/L
- Meticulous monitoring flowsheet: vital signs, glucose (hourly), electrolytes (2-hourly initially), fluid intake/output
2. Fluid Resuscitation (FIRST PRIORITY)
- Phase 1 (First hour): 0.9% Normal Saline (isotonic) - 1 L over first hour (or faster if hypotensive/shocked: boluses of 20 mL/kg until SBP >80 mmHg)
- Phase 2 (Next 2-3 hours): Continue 0.9% NS at 250-500 mL/hour (2-4 L total in first 2-4 hours)
- Phase 3 (After initial resuscitation): Switch to 0.45% NS at 125-250 mL/hour to address the free water deficit
- When glucose falls to ≤250-300 mg/dL: Add dextrose - switch to D5W/0.45% NS to prevent hypoglycemia while continuing insulin to clear ketones
- Total fluid deficit = approximately 70-90 mL/kg; replace over 24-48 hours
Note on this patient: Na⁺ is 120 mEq/L (pseudohyponatremia). Do NOT correct sodium faster than 10-12 mEq/L per 24 hours to avoid cerebral edema/osmotic demyelination.
3. Potassium Replacement (CRITICAL - must precede insulin)
⚠ This patient has K⁺ 6.8 mEq/L - do NOT give K⁺ yet. Start insulin cautiously after confirming urine output.
| Serum K⁺ | Action |
|---|---|
| <3.3 mEq/L | Replete to >3.3 mEq/L BEFORE starting insulin (40 mEq/hr IV) |
| 3.3-5.5 mEq/L | Add 20-40 mEq K⁺ per liter of IV fluid to maintain levels |
| >5.5 mEq/L (this patient) | Do NOT supplement K⁺; reassess every 1-2 hours as K⁺ will fall rapidly with insulin therapy |
4. Insulin Therapy
Do not start insulin until K⁺ >3.3 mEq/L (risk of fatal hypokalemia).
- No IV bolus (no longer recommended)
- Regular insulin IV infusion: 0.1 units/kg/hour (e.g., 7 units/hour for 70 kg patient)
- Target glucose reduction rate: 50-70 mg/dL/hour
- Once glucose reaches 250 mg/dL: reduce insulin to 0.05 units/kg/hour AND add dextrose to IV fluids
- Continue insulin infusion until ketonemia resolves (not just until glucose normalizes) - pH >7.3, HCO₃ >15 mEq/L, anion gap normalized
- Transition to subcutaneous insulin: Once patient can eat, overlapping SC insulin with IV for 1-2 hours before stopping the infusion (to prevent rebound ketosis)
5. Treat the Precipitant
- This patient: Treat the underlying infection (source workup: blood cultures, urine culture, CXR for pneumonia, wound cultures)
- Start empirical broad-spectrum antibiotics if sepsis suspected
- ECG to rule out acute MI as precipitant
- Serum amylase/lipase if abdominal pain prominent (pancreatitis)
6. Bicarbonate - Controversial, Rarely Used
- Generally NOT recommended in DKA (even with severe acidosis pH 7.1)
- Rationale against: rapid correction paradoxically worsens CNS acidosis, causes hypokalemia, impairs oxygen delivery (leftward Hb-O₂ curve shift)
- Possible consideration: If pH <6.9 and hemodynamic compromise (given as 50-100 mEq NaHCO₃ in 200-400 mL sterile water over 1-2 hours with potassium supplementation)
7. Phosphate
- Routine replacement not recommended (no clear clinical benefit shown)
- Replace if measured serum phosphate <1.0 mEq/L (use potassium phosphate to also address K⁺ deficit)
8. Monitoring During Treatment
- Glucose: hourly (bedside glucometry)
- Electrolytes, BUN, creatinine, venous pH: every 2-4 hours
- Urine output: hourly via catheter
- Vital signs: every 30-60 minutes
- ECG monitoring (for K⁺-related arrhythmias - this patient has K⁺ 6.8 → risk of peaked T waves, widened QRS, VF)
- Watch for complications of treatment: hypoglycemia, hypokalemia, cerebral edema (especially in children), hyperchloremic acidosis
d. Advice to Avoid Recurrence
1. "Sick Day Rules" - Most Important Preventive Strategy
- Never stop insulin during illness, even if unable to eat - illness increases insulin requirements
- Monitor blood glucose more frequently during any illness (every 2-4 hours)
- Monitor urine or blood ketones when glucose >250 mg/dL
- Increase fluid intake during illness to prevent dehydration
- Contact the doctor or go to the emergency department if glucose consistently >300 mg/dL, moderate/large ketones, vomiting prevents oral intake, or mental status changes
2. Strict Medication Adherence
- Never skip insulin doses - missing doses is the second most common cause of DKA
- Review insulin storage and technique (improper storage inactivates insulin)
- Ensure caregiver/family understands insulin administration
- Use an insulin pen/device if compliance is an issue
3. Blood Glucose Self-Monitoring
- Regular home glucose monitoring - at least twice daily; more during illness
- HbA1c target <7% (individualized for elderly patients, <8% may be acceptable)
- Know the signs of hyperglycemia (polyuria, polydipsia, fatigue) and act early
4. Prevent and Treat Infections Promptly
- Annual influenza vaccination
- Pneumococcal vaccine (particularly important in elderly diabetics)
- COVID-19 vaccination
- Good foot care - daily foot inspection, appropriate footwear; prompt treatment of any infections
- Maintain good oral hygiene (dental infections cause bacteremia)
- Treat any infection early and aggressively - do not delay seeking care
5. Regular Medical Follow-up
- Regular HbA1c checks (every 3-6 months)
- Annual eye exam (retinopathy), renal function (nephropathy), foot exam, neuropathy screening
- Medication review - check if any new drugs (e.g., corticosteroids, SGLT-2 inhibitors) increase DKA risk
- Dietitian referral for consistent carbohydrate intake planning
6. Patient and Family Education
- Educate family members/caregivers about signs of DKA (altered sensorium, Kussmaul breathing, acetone breath, vomiting)
- Provide a written sick-day action plan
- Wear a medical alert bracelet identifying diabetes
7. Special Consideration for SGLT-2 Inhibitors
- If this patient is on empagliflozin/dapagliflozin/canagliflozin: counsel about risk of euglycemic DKA (normal glucose with ketones) - may need to withhold during illness, surgery, or fasting
- Euglycemic DKA is easy to miss because glucose appears "normal"
8. Address Modifiable Risk Factors
- Optimal glycemic control (reduces threshold for DKA)
- Avoid alcohol excess (promotes ketosis, impairs gluconeogenesis, causes missed insulin doses)
- Manage stress (physical and psychological stress raises counter-regulatory hormones)
- Goldman-Cecil Medicine, pp. 2483-2484 (DKA: Biochemistry, Diagnosis, Treatment)
- Rosen's Emergency Medicine, pp. 2542-2544 (Pathophysiology, Clinical Features, Management Tables)
- Rosen's Emergency Medicine, Table 115.3: Average Fluid and Electrolyte Deficits in Severe DKA
- Rosen's Emergency Medicine, Table 115.4: Typical Laboratory Values in DKA vs. HHS
Raju, a 26-year-old male, presents with shortness of breath and wheeze with a history of seasonal variation. a. What is the diagnosis? b. Enumerate the clinical features. c. Investigations and their relevance. d. Discuss management and preventive measures.
"asthma"[MeSH Terms] AND "management"[MeSH Terms]
"asthma"[MeSH Terms] AND "guideline"[pt]
Bronchial Asthma
a. Diagnosis
- Episodic shortness of breath and wheeze - hallmark of reversible airflow obstruction
- Seasonal variation - characteristic of allergen-driven (atopic) asthma; symptoms worsen during pollen seasons (spring/autumn), cold weather, or periods of high dust/mold exposure
- Young age (26 years) - asthma most commonly begins before age 25; atopic asthma predominates in young males
Types of Asthma
| Type | Features | This Patient |
|---|---|---|
| Atopic/Extrinsic/Allergic | IgE-mediated, starts in childhood/young adulthood, seasonal, associated with eczema/rhinitis, positive skin tests | Most likely |
| Non-atopic/Intrinsic | Starts in adulthood, no allergies, often with nasal polyps, worse prognosis | Less likely |
| Occupational | Triggered by workplace exposures | Consider if seasonal = work-related |
| Exercise-induced | Precipitated by physical exertion | May coexist |
| Aspirin-sensitive | Triad: asthma, nasal polyps, aspirin intolerance | Less likely here |
b. Clinical Features
Symptoms
- Wheeze - high-pitched musical sound, predominantly expiratory, caused by turbulent airflow through narrowed airways
- Dyspnoea (Shortness of breath) - episodic, often worse at night and early morning (nocturnal and early morning dipping of peak flow)
- Cough - often dry, hacking; may be the only symptom in "cough-variant asthma"
- Chest tightness - a feeling of constriction, often precedes wheeze
- Symptoms are episodic with symptom-free intervals between attacks
- Seasonal variation - worse in spring (tree pollen), summer (grass pollen), or autumn (mould spores, dust mites)
- Nocturnal worsening - characteristic; due to circadian fall in cortisol, increased parasympathetic tone, reduced mucociliary clearance during sleep
- Symptoms triggered by: exercise, cold air, upper respiratory infections (viral), allergen exposure, irritants (smoke, fumes), emotional stress, NSAIDs/beta-blockers
Signs on Physical Examination
- Tachypnoea - increased respiratory rate
- Tachycardia
- Use of accessory muscles of respiration (sternocleidomastoid, scalenes)
- Hyperinflated chest (barrel-shaped chest in chronic/severe asthma) - due to air trapping
- Intercostal and subcostal recession (in children)
- Reduced chest expansion
- Hyperresonance on percussion (air trapping)
- Prolonged expiratory phase with widespread bilateral expiratory wheeze on auscultation
- Reduced air entry in severe bronchospasm
- Examination may be completely normal (a key feature of asthma)
- May have coexisting features of atopy: allergic rhinitis, eczema, nasal polyps
Signs of a Severe/Life-Threatening Attack (Status Asthmaticus)
| Feature | Significance |
|---|---|
| Silent chest (no wheeze) | Severe airflow obstruction - air movement too poor to generate wheeze |
| Cyanosis | Severe hypoxia |
| Pulsus paradoxus >10 mmHg | Severe obstruction causing exaggerated intrathoracic pressure swings |
| Inability to speak in sentences | Severe respiratory distress |
| Exhaustion, altered consciousness | Near-fatal attack |
| Peak flow <33% predicted | Life-threatening |
Atopic Features (Support Allergic Asthma Diagnosis)
- Personal or family history of eczema, allergic rhinitis (hay fever), or food allergy ("atopic triad")
- Peripheral blood eosinophilia
- Elevated total serum IgE
c. Investigations and Their Relevance
1. Spirometry (Pulmonary Function Tests) - MOST IMPORTANT
- FEV₁ (Forced Expiratory Volume in 1 sec) - reduced
- FVC (Forced Vital Capacity) - normal or mildly reduced
- FEV₁/FVC ratio - reduced (<0.7 or <70% = obstructive pattern)
- Reversibility test: Give a short-acting bronchodilator (salbutamol 400 µg) → repeat spirometry after 15-20 minutes
- ≥12% AND ≥200 mL increase in FEV₁ = significant reversibility = confirms asthma
2. Peak Expiratory Flow Rate (PEFR)
- Measured with a portable peak flow meter - simple, cheap, repeatable
- Diurnal variation >20% (morning dip pattern) is characteristic of asthma
- Normal PEFR: 350-550 L/min (varies with age, sex, height)
- Used to monitor response to treatment and classify severity
- Relevance: Objective home monitoring; essential for the "traffic light" asthma action plan
3. Bronchoprovocation / Methacholine Challenge Test
- Used when spirometry is normal but asthma is clinically suspected
- Inhaled methacholine (or histamine) is given in increasing doses → FEV₁ is measured
- PC₂₀ = concentration causing 20% fall in FEV₁; PC₂₀ <8 mg/mL = airway hyperresponsiveness (positive for asthma)
- Relevance: Demonstrates bronchial hyperresponsiveness (BHR) - the defining physiological abnormality of asthma. Highly sensitive (negative test effectively rules out asthma).
4. Allergy Testing
- Skin prick tests (allergen extracts applied to skin, wheal and flare response measured): identifies specific IgE-mediated sensitivities (house dust mite, grass pollen, animal dander, moulds)
- Serum specific IgE (RAST/ImmunoCAP): measures IgE antibodies against specific allergens
- Total serum IgE (elevated in atopic asthma)
- Relevance: Identifies specific triggers; guides allergen avoidance and eligibility for immunotherapy (desensitization) and biologics (e.g., omalizumab requires elevated IgE or specific IgE)
5. Full Blood Count
- Eosinophilia (>0.3 × 10⁹/L in blood): supports atopic asthma; elevated eosinophils are now a key biomarker guiding biologic therapy (mepolizumab, benralizumab)
- Relevance: Distinguishes eosinophilic vs. non-eosinophilic asthma, guides biologic selection
6. Exhaled Nitric Oxide (FeNO)
- Fractional exhaled nitric oxide - a marker of eosinophilic airway inflammation
- FeNO >40 ppb = elevated = eosinophilic inflammation likely = suggests ICS-responsive asthma
- Relevance: Non-invasive biomarker; predicts steroid responsiveness; guides therapy titration; monitors airway inflammation
7. Chest X-Ray
- Usually normal between attacks in uncomplicated asthma
- During an acute severe attack: hyperinflation (flattened diaphragms, increased AP diameter, hyperlucency)
- Rules out differentials: pneumonia, pneumothorax, foreign body, cardiac failure
- Relevance: Not diagnostic for asthma but excludes alternative diagnoses and complications
8. Arterial Blood Gas (ABG)
- In acute severe asthma: initially shows respiratory alkalosis (hypocapnia - PCO₂ <35 mmHg) due to hyperventilation
- Normal or rising PCO₂ during an acute attack is an ominous sign of respiratory fatigue → impending respiratory failure
- Hypoxaemia (PaO₂ <60 mmHg) indicates severe disease
- Relevance: Guides ICU admission and ventilatory support decisions in acute severe asthma
9. Sputum Analysis
- Eosinophilic sputum (>3% eosinophils): supports eosinophilic asthma
- Charcot-Leyden crystals (from eosinophil breakdown) and Curschmann's spirals (mucus plugs) are classic but non-specific findings in asthma sputum
- Relevance: Identifies inflammatory phenotype; guides anti-eosinophilic therapy
10. Skin tests / IgE Panel Summary of Relevance
| Investigation | What It Shows | Clinical Use |
|---|---|---|
| Spirometry + reversibility | Obstructive pattern + ≥12%/200mL FEV₁ reversal | Diagnosis confirmation |
| PEFR diary | Diurnal variation >20% | Diagnosis + monitoring |
| Methacholine challenge | BHR (PC₂₀ <8 mg/mL) | Diagnosis in normal spirometry |
| Skin prick / specific IgE | Specific allergic sensitization | Trigger identification; biologic eligibility |
| FeNO | Eosinophilic inflammation | Phenotyping; steroid response prediction |
| Blood eosinophils | Eosinophilic vs. non-eosinophilic | Biologic selection |
| CXR | Hyperinflation; exclude other diagnoses | Baseline; acute assessment |
| ABG | Degree of hypoxia/hypercapnia | Severity assessment in acute attack |
d. Management and Preventive Measures
GINA (Global Initiative for Asthma) Step-Up Approach
Step 1: As-Needed Low-Dose ICS-Formoterol (Mild Asthma)
- Previous approach (still widely used): SABA (short-acting beta-2 agonist) as reliever, e.g., salbutamol (albuterol) 100 µg 1-2 puffs PRN
- ICS-formoterol as reliever reduces exacerbation risk compared to SABA alone
Step 2: Low-Dose ICS Daily + As-Needed Reliever
- Preferred controller: Low-dose ICS (e.g., budesonide 200-400 µg/day or beclomethasone 200-500 µg/day)
- ICS is the single most effective preventer in asthma - reduces inflammation, exacerbations, and mortality
- Alternative: Leukotriene receptor antagonist (LTRA, e.g., montelukast 10 mg daily) - less effective than ICS but useful in patients who cannot use inhalers or have concurrent allergic rhinitis
Step 3: Low-Dose ICS + Long-Acting Beta-2 Agonist (LABA)
- Preferred: Low-dose ICS + LABA combination inhaler (e.g., salmeterol/fluticasone, formoterol/budesonide)
- LABA must NEVER be used without ICS in asthma (risk of severe exacerbations)
- Alternative: Medium-dose ICS alone
- Add-on: LTRA (montelukast), tiotropium (LAMA - long-acting muscarinic antagonist)
Step 4: Medium-High Dose ICS + LABA
- Medium or high-dose ICS/LABA combination
- Add tiotropium (LAMA) - approved add-on in adults with ≥1 exacerbation/year
- Consider referral to specialist
Step 5: High-Dose ICS + LABA + Specialist Add-on Therapy (Severe Asthma)
- Biologic therapies (targeted therapy based on phenotype):
| Biologic | Target | Indication |
|---|---|---|
| Omalizumab (anti-IgE) | IgE | Moderate-severe allergic asthma, elevated total/specific IgE |
| Mepolizumab / Reslizumab (anti-IL-5) | IL-5 | Severe eosinophilic asthma, blood eos ≥300 cells/µL |
| Benralizumab (anti-IL-5Rα) | IL-5 receptor | Severe eosinophilic asthma |
| Dupilumab (anti-IL-4Rα) | IL-4/IL-13 | Severe type-2 high asthma |
| Tezepelumab (anti-TSLP) | TSLP (alarmin) | Severe asthma regardless of phenotype |
- Low-dose oral corticosteroids (last resort - significant side effects)
- Bronchial thermoplasty (interventional bronchoscopy to reduce airway smooth muscle mass)
Management of an Acute Asthma Attack
| Moderate | Severe | Life-Threatening |
|---|---|---|
| Increasing symptoms | Can't complete sentences | Silent chest |
| PEFR 50-75% | PEFR 33-50% | PEFR <33% |
| No features of severe | RR >25, HR >110 | Cyanosis, exhaustion, hypotension |
- Oxygen: High-flow O₂ to maintain SpO₂ 94-98%
- SABA (Salbutamol): 2.5-5 mg nebulized every 20 minutes for first hour, then as needed; or salbutamol 4-8 puffs via spacer every 20 min
- Ipratropium bromide (SAMA): 0.5 mg nebulized - add in severe/life-threatening attack; synergistic bronchodilation
- Systemic corticosteroids: Prednisolone 40-50 mg orally (or IV hydrocortisone 100 mg if unable to swallow) - reduces airway inflammation, prevents relapse; continue for 5-7 days
- IV Magnesium sulphate: 1.2-2 g IV over 20 minutes - for severe/life-threatening attack not responding to initial treatment; causes smooth muscle relaxation
- IV Aminophylline: Second-line, now rarely used; risk of toxicity (theophylline-containing)
- Heliox (helium-oxygen mixture): reduces turbulent airflow resistance - specialist use
- Non-invasive or invasive ventilation: If impending respiratory failure (rising PCO₂, exhaustion, altered consciousness)
Drug Summary
| Drug Class | Examples | Route | Role |
|---|---|---|---|
| SABA | Salbutamol, terbutaline | Inhaled | Reliever - acute bronchospasm |
| LABA | Salmeterol, formoterol | Inhaled (with ICS) | Controller - long-term prevention |
| ICS | Budesonide, fluticasone, beclomethasone | Inhaled | Controller - anti-inflammatory mainstay |
| LTRA | Montelukast | Oral | Add-on controller; useful in allergic rhinitis |
| LAMA | Tiotropium | Inhaled | Add-on controller (Step 4+) |
| Methylxanthines | Theophylline | Oral/IV | Add-on; narrow therapeutic window |
| SAMA | Ipratropium bromide | Inhaled | Acute attacks (add-on to SABA) |
| Corticosteroids | Prednisolone | Oral/IV | Acute attacks; oral maintenance in severe asthma |
| Biologics | Omalizumab, mepolizumab | SC injection | Severe uncontrolled asthma |
Preventive Measures
- Breastfeeding for at least 4-6 months (reduces atopic sensitization)
- Avoidance of tobacco smoke exposure in infancy
- Farm/rural upbringing and early microbial exposure (the "hygiene hypothesis" - contact with animals and diverse microbiome reduces atopy risk)
- Avoid indoor dampness and mould exposure
- House dust mite (HDM) reduction: Allergen-proof mattress/pillow covers, wash bedding in hot water (>60°C) weekly, remove carpets and soft furnishings, reduce indoor humidity
- Pollen avoidance: Keep windows closed during high pollen count days, check pollen forecasts, wear sunglasses outdoors, shower after outdoor activities
- Pet allergen avoidance: Avoid pet ownership if sensitized to cat/dog dander; if unavoidable, exclude pets from bedrooms
- Avoid tobacco smoke (both active smoking and passive exposure) - major irritant and risk factor for severity
- Avoid occupational sensitizers if applicable (isocyanates, flour dust, latex)
- Avoid NSAIDs and aspirin if aspirin-sensitive asthma (check for nasal polyps)
- Beta-blockers: Avoid (even topical ophthalmic preparations) - can precipitate severe bronchospasm
- Avoid cold air exposure - use a scarf over the nose/mouth in winter
- Prevent viral URTIs: Annual influenza vaccination; pneumococcal vaccine; hand hygiene
- Subcutaneous immunotherapy (SCIT) or sublingual immunotherapy (SLIT) for house dust mite or pollen-sensitive allergic asthma
- Gradually increasing doses of allergen → immune tolerance
- Reduces both asthma AND rhinitis symptoms; may modify disease progression
- Most effective for mild-moderate allergic asthma with clear single allergen sensitivity
- Weight reduction if obese - obesity is an independent risk factor for severe/difficult-to-control asthma
- Regular exercise (with pre-treatment with SABA if exercise-induced) - improves cardiovascular fitness, reduces BMI, improves asthma control
- Breathing exercises (Buteyko technique, physiotherapy-led) - reduces reliever use and improves quality of life
- Stress management - psychological stress triggers attacks; CBT or mindfulness may help
- Avoid air pollution exposure (particularly on high-pollution days)
- Avoid occupational exposure to irritants
- Provide a written Personalised Asthma Action Plan (PAAP) - "traffic light" system based on PEFR and symptoms (green/amber/red zones with instructions for each)
- Teach correct inhaler technique (40-80% of patients use inhalers incorrectly)
- PEFR diary to monitor control and detect deterioration early
- Regular scheduled follow-up (not just when symptomatic)
- Educate about recognising and responding to worsening control before a crisis develops
- Goldman-Cecil Medicine, pp. 879-880+ (Definition, Epidemiology, Pathobiology, Types, Treatment)
- Murray & Nadel's Textbook of Respiratory Medicine (Pathophysiology, Impairment Rating, Clinical Features)
- Textbook of Family Medicine, 9e (Asthma in clinical practice)
- GINA 2024 Guidelines (Step-wise management framework)
A 53-year-old male with long-standing Type 2 Diabetes Mellitus, hypertension, hyperlipidemia, smoking history, retinopathy, edema, pruritus, and abnormal laboratory findings. a. Diagnose the case. b. Discuss the pathogenesis and histological classification. c. Enumerate possible causes of anemia in this case. d. Enumerate major risk factors for progression (5 points). e. Enumerate specific goals for prevention of the disease. f. Explain guidelines regarding metformin and ACE-inhibitor use in this case.
"diabetic nephropathy"[MeSH Terms] AND "progression"[MeSH Terms]
Diabetic Nephropathy (Diabetic Kidney Disease - DKD)
a. Diagnosis
| Clinical Feature | Significance |
|---|---|
| 53-year-old, long-standing T2DM | Primary cause of nephropathy; 30-40% of T2DM patients develop nephropathy |
| Hypertension | Both a cause and consequence of diabetic nephropathy; accelerates progression |
| Hyperlipidemia | Independent risk factor for nephropathy progression and cardiovascular disease |
| Smoking history | Independent risk factor for nephropathy progression |
| Retinopathy | Key correlating microvascular complication - >60% of T2DM nephropathy patients have retinopathy; presence strongly suggests nephropathy is diabetic in origin |
| Edema | Hypoalbuminemia from proteinuria (nephrotic-range) + reduced GFR → sodium and water retention |
| Pruritus | Uremia (accumulation of uremic toxins as GFR falls) → uremic pruritus |
| Abnormal lab findings | Elevated creatinine/BUN, proteinuria, anemia of CKD, hyperkalemia, metabolic acidosis |
b. Pathogenesis and Histological Classification
Pathogenesis
- Hyperglycemia → upregulation of SGLT1 and SGLT2 in the proximal tubule → increased Na⁺/glucose reabsorption → decreased Na⁺ delivery to the macula densa → macula densa cannot "sense" adequate NaCl → afferent arteriolar dilation + RAAS activation → glomerular hyperfiltration (GFR transiently elevated)
- Increased intraglomerular pressure damages the capillary wall and podocytes
- Persistent hyperglycemia → non-enzymatic glycation of proteins and lipids → AGEs accumulate in glomerular basement membrane (GBM), mesangium, and vessel walls
- AGEs cross-link matrix proteins, increase GBM permeability, stimulate TGF-β production, promote mesangial expansion
- Hyperglycemia + glomerular hyperfiltration → angiotensin II excess
- Angiotensin II → preferential efferent arteriolar constriction → further increases intraglomerular pressure → promotes TGF-β production → matrix accumulation and fibrosis
- ACE gene D allele polymorphism is associated with higher ACE activity and susceptibility to ESKD
- TGF-β (transforming growth factor-β) is the key profibrotic cytokine
- Stimulates synthesis of collagen types III and IV and fibronectin → subepithelial and mesangial matrix expansion → glomerulosclerosis
- Hyperglycemia → reactive oxygen species (ROS) generation → endothelial dysfunction, podocyte injury
- Pro-inflammatory cytokines (IL-6, TNF-α, VCAM-1, ICAM-1) → leukocyte infiltration → tubulointerstitial nephritis
- Normal GBM contains heparan sulfate proteoglycans (negatively charged) → repel albumin (also negatively charged)
- Diabetes → loss of heparan sulfate moieties → albumin crosses freely → proteinuria
- Progressive GBM thickening despite loss of charge barrier
- Podocyte injury and detachment → effacement of foot processes → loss of size-selective filtration barrier → massive proteinuria in late disease
Natural History (Mogensen Staging - Type 1 DM, applicable as framework for T2DM)
| Stage | Features | Timing |
|---|---|---|
| Stage 1 - Glomerular Hypertrophy & Hyperfiltration | GFR elevated (↑20-40%), kidney enlargement, no proteinuria | At onset of diabetes |
| Stage 2 - Silent/Latent | GBM thickening, mesangial expansion, normoalbuminuria or exercise-induced microalbuminuria; GFR still elevated | 2-5 years |
| Stage 3 - Incipient Nephropathy | Microalbuminuria (30-300 mg/24h or 30-300 mg/g creatinine); GFR still normal or mildly reduced | 5-15 years |
| Stage 4 - Overt/Clinical Nephropathy | Macroproteinuria (>300 mg/24h); declining GFR (~10 mL/min/year); hypertension; edema | 10-20 years |
| Stage 5 - End-Stage Renal Disease | GFR <15 mL/min/1.73 m²; uremia; dialysis or transplant needed | 15-25 years |
Note: In T2DM, onset is uncertain - patients may present at any stage, including advanced nephropathy, at initial diabetes diagnosis.
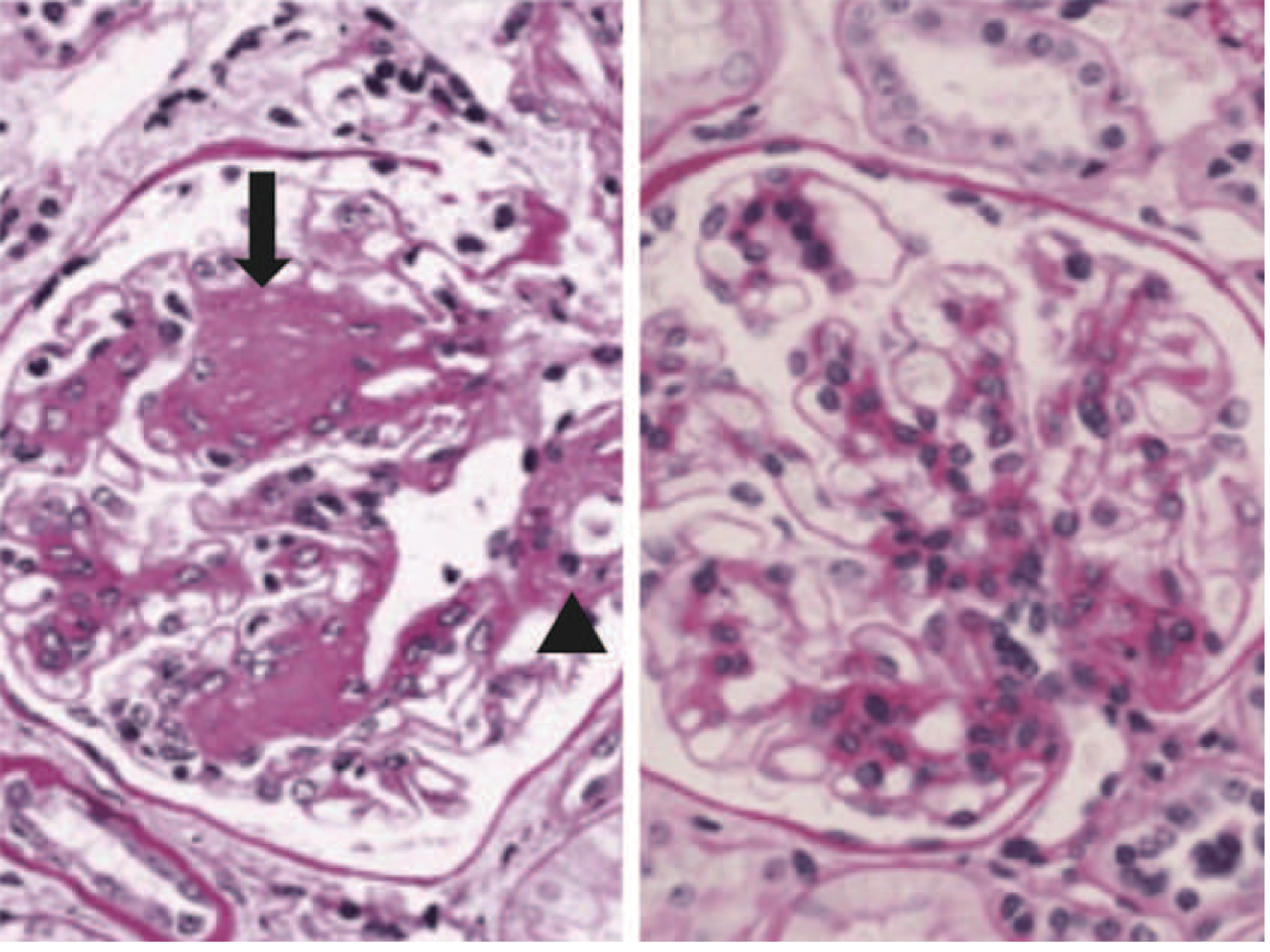
Histological Classification (Robbins / Harrison's)
- Earliest morphological change, detectable by EM within years of diabetes onset
- Uniform thickening along entire length of capillary loops
- Correlates poorly with clinical manifestations in early disease
- Increase in mesangial matrix with mesangial cell proliferation and GBM thickening
- Found in most patients with >10 years of diabetes
- More common with hypertension and older age
- When severe, produces the nephrotic syndrome (proteinuria, hypoalbuminemia, edema)
- Ball-like, laminated, eosinophilic, PAS-positive nodules in the periphery of the glomerulus (mesangial nodules)
- Located in the mesangium at the periphery of the glomerulus
- Found in 15-30% of patients with long-term diabetes
- Virtually pathognomonic of diabetes (in contrast to diffuse sclerosis, which is less specific)
- Electron microscopy: no immune deposits (distinguishes from membranoproliferative GN)
- Immunofluorescence: non-specific IgG linear deposition or complement without immune deposits
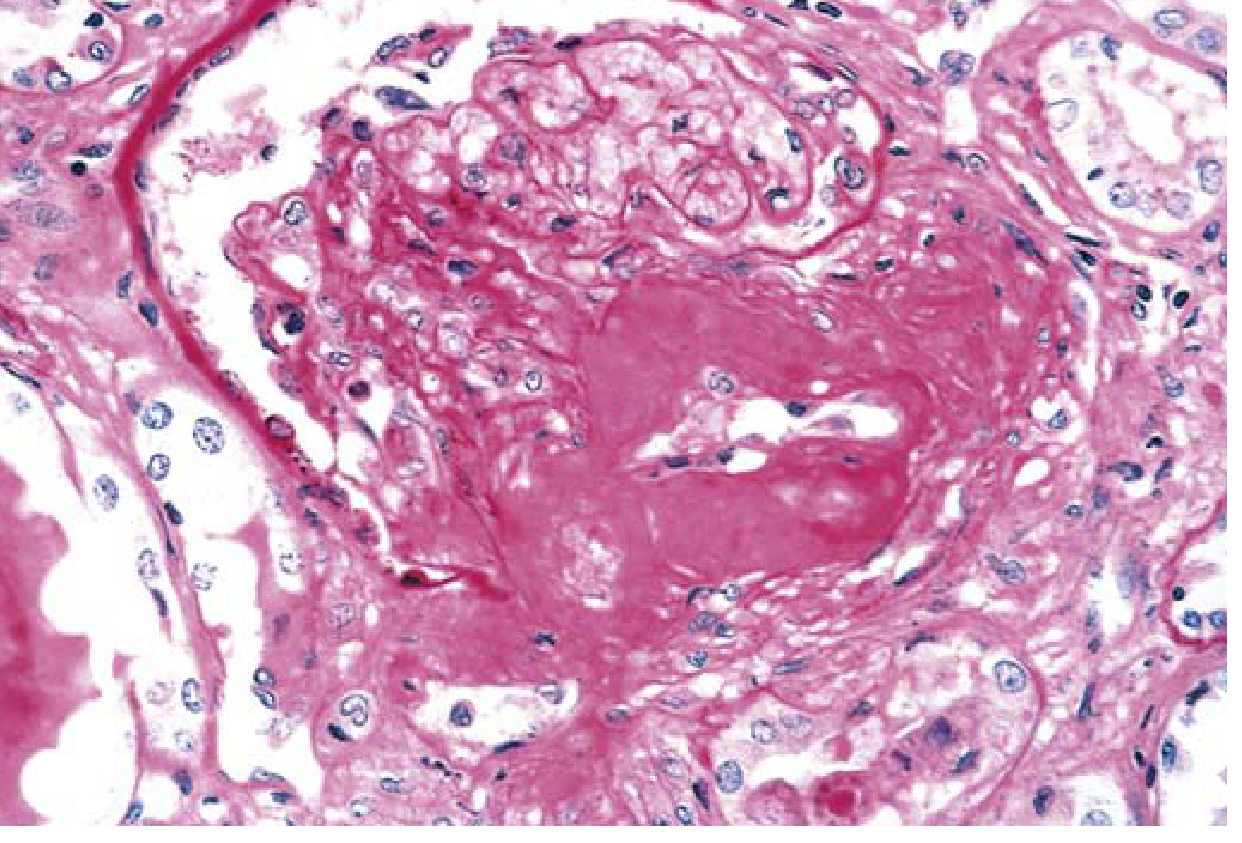
- Hyaline arteriolosclerosis of BOTH afferent AND efferent arterioles - efferent involvement is virtually unique to diabetes (non-diabetic hypertension affects only afferent)
- Renal atherosclerosis (macrovascular disease)
- Leads to ischemia and nephrosclerosis (finely granular cortical surface)
- Tubular atrophy and interstitial fibrosis (correlates best with GFR decline)
- "Armanni-Ebstein lesion" - glycogen accumulation in tubular cells (characteristic but not pathognomonic)
- Increased susceptibility to pyelonephritis
- Necrotizing papillitis (papillary necrosis) is much more prevalent in diabetics than non-diabetics
c. Causes of Anemia in This Patient
- Failing kidneys produce insufficient erythropoietin (EPO) (made by peritubular fibroblasts in the renal cortex) → reduced RBC production by bone marrow
- This is the dominant cause of anemia in CKD; appears early (GFR <60 mL/min) and worsens as GFR falls
- Absolute iron deficiency: poor dietary intake, reduced intestinal iron absorption (elevated hepcidin in CKD blocks ferroportin)
- Functional iron deficiency: inadequate iron mobilization for erythropoiesis even with normal iron stores
- GI blood loss (uremic gastritis, peptic ulcer disease)
- Uremia itself is a pro-inflammatory state → elevated IL-6, TNF-α → upregulates hepcidin → sequesters iron in macrophages → reduced iron availability for erythropoiesis
- Uremic toxins shorten RBC lifespan
- Uremic environment damages the RBC membrane (eryptosis)
- Uremic toxins directly suppress bone marrow erythroid precursors
- Secondary hyperparathyroidism (common in advanced CKD) can cause bone marrow fibrosis (osteitis fibrosa cystica), reducing erythropoiesis
- Folate deficiency (reduced dietary intake, dialysis losses)
- Vitamin B12 deficiency (altered absorption)
- Malnutrition from uremic anorexia
- If on hemodialysis: blood loss in dialyzer tubing and lines
- Frequent blood sampling for investigations
- Aluminum-containing phosphate binders → microcytic, hypochromic anemia resistant to EPO therapy (less common now)
d. Major Risk Factors for Progression (5 Points)
-
Hyperglycemia / Poor Glycemic Control
- Elevated HbA1c is the strongest modifiable metabolic risk factor
- Each 1% rise in HbA1c accelerates microalbuminuria progression
- Duration of diabetes correlates directly with nephropathy progression
-
Hypertension
- Systemic hypertension directly transmits to glomerular capillaries (especially in diabetics who have impaired autoregulation)
- Accelerates glomerulosclerosis, tubulointerstitial fibrosis, and vascular disease
- Blood pressure >130/80 mmHg strongly predicts faster GFR decline
-
Proteinuria / Albuminuria Level
- Albuminuria is the single most important predictor of faster GFR decline
- Macroalbuminuria (>300 mg/g) predicts ESKD far more than microalbuminuria
- Proteinuria itself causes tubular toxicity (filtered proteins activate tubular cells → fibrosis)
-
Dyslipidemia
- Elevated LDL, triglycerides, and reduced HDL accelerate glomerulosclerosis
- Lipid deposits in mesangial cells → foam cell formation → matrix expansion
- Dyslipidemia also promotes cardiovascular mortality (the leading cause of death in diabetic nephropathy)
-
Smoking
- Independent risk factor for onset and progression of diabetic nephropathy
- Worsens glomerular hyperfiltration, endothelial dysfunction, and atherosclerosis
- Smoking cessation significantly slows nephropathy progression
e. Specific Goals for Prevention of Diabetic Nephropathy
Primary Prevention (Preventing Onset - Stage 1 to 3)
-
Glycemic Control Target
- HbA1c goal: <7.0% (ADA) in most patients with T2DM
- More stringent targets (<6.5%) may benefit patients with early nephropathy and low hypoglycemia risk
- Improved glycemic control reduces the rate at which microalbuminuria appears and progresses in both T1DM and T2DM
- SGLT2 inhibitors and GLP-1 receptor agonists (liraglutide, semaglutide) provide renal-protective benefits beyond glycemic control
-
Blood Pressure Control
- Target: <130/80 mmHg in patients with diabetes (ADA/Harrison's)
- Most patients with diabetic nephropathy require ≥3 antihypertensive medications
- BP control reduces both cardiovascular and kidney adverse events
-
RAAS Blockade (ACE inhibitor or ARB)
- Indicated once microalbuminuria is detected (UACR ≥30 mg/g)
- Reduces albuminuria and slows GFR decline independent of blood pressure reduction
- Goal: Reduce UACR by ≥30% in patients with UACR >300 mg/g
-
SGLT2 Inhibitor (e.g., empagliflozin, dapagliflozin, canagliflozin)
- In T2DM with eGFR >20 mL/min/1.73 m², addition of SGLT2i to ACEi/ARB reduces risk of kidney failure and cardiovascular events
- Dapagliflozin (DAPA-CKD trial) reduces CKD progression and cardiovascular death even without T2DM
-
Lipid Management
- LDL-C target: <70 mg/dL (high cardiovascular risk)
- Statin therapy indicated for all T2DM patients with nephropathy
- Dyslipidemia should be treated aggressively (Harrison's)
-
Smoking Cessation
- Absolute requirement; smoking cessation slows nephropathy progression
-
Screening and Monitoring
- T2DM: Test urine albumin-to-creatinine ratio (UACR) and eGFR at diagnosis, then annually
- Detect microalbuminuria early (UACR 30-300 mg/g) → intervene before overt nephropathy
-
Dietary Protein Restriction
- ADA recommends 0.8 g/kg/day of protein in patients with diabetic kidney disease (not lower - excessive restriction risks malnutrition)
- Reduces intraglomerular pressure and proteinuria
-
Finerenone (Nonsteroidal Mineralocorticoid Receptor Antagonist)
- In patients with T2DM and nephropathy already on ACEi or ARB: finerenone (FIDELIO-DKD trial) improved cardiovascular and kidney outcomes
- Reduces residual albuminuria; requires monitoring of serum potassium
-
Nephrology Referral
- eGFR <30 mL/min/1.73 m²
- Albuminuria >300 mg/g creatinine
- Atypical features (hematuria, rapid GFR decline, absence of retinopathy)
f. Guidelines Regarding Metformin and ACE Inhibitor Use
Metformin in Diabetic Nephropathy
| eGFR (mL/min/1.73 m²) | Recommendation |
|---|---|
| ≥60 | Safe to use; no dose restriction |
| 45-60 | Continue with caution; check eGFR more frequently (every 3-6 months) |
| 30-45 | Use with caution; if already on metformin, consider risk/benefit; reduce dose |
| <30 | Contraindicated (absolute contraindication - risk of lactic acidosis) |
| Any eGFR | Hold metformin perioperatively, before contrast media administration, and during acute illness (dehydration, sepsis, hypoxia) |
- The Harriet Lane Handbook confirms: metformin is contraindicated in severe renal impairment (eGFR <30 mL/min/1.73 m²) due to risk of lactic acidosis
- Sulfonylureas are similarly contraindicated in advanced renal insufficiency (risk of prolonged hypoglycemia)
- SGLT2 inhibitors are not effective with eGFR <20 mL/min/1.73 m²
ACE Inhibitor (or ARB) in Diabetic Nephropathy
- Block angiotensin II → preferential efferent arteriolar dilation → reduces intraglomerular hypertension
- Reduce TGF-β production → less mesangial fibrosis
- Reduce proteinuria (independent of systemic BP lowering) → slow GFR decline
- Reduce aldosterone → reduce sodium retention and fibrosis
| Clinical Situation | Recommendation |
|---|---|
| Microalbuminuria (UACR 30-300 mg/g) | ACEi or ARB recommended - slows progression to overt nephropathy |
| Macroalbuminuria (UACR >300 mg/g) + CKD | ACEi or ARB strongly recommended - first-line antihypertensive AND renoprotective |
| No albuminuria, no hypertension | RAAS inhibitors do NOT prevent development of nephropathy - not indicated |
| ACEi cough | Switch to ARB (equivalent efficacy) |
| ACEi + ARB combination | NOT recommended - no additional benefit; increases risk of hyperkalemia and acute kidney injury |
| Pregnancy (2nd/3rd trimester) | ACEi/ARB contraindicated (fetotoxic: skull hypoplasia, renal tubular dysplasia, IUGR) |
- Serum creatinine and potassium: check 1-2 weeks after starting and after each dose increase
- A rise in serum creatinine up to 30% is acceptable and expected (loss of efferent tone reduces GFR slightly) - do NOT stop the drug
- If creatinine rises >30% → evaluate for bilateral renal artery stenosis, severe volume depletion
- Target: reduce UACR by ≥30%
- Hyperkalemia (K⁺ >5.5 mEq/L) → dose reduction or switch agents; use finerenone cautiously
- ACEi/ARB should be continued as long as eGFR >15-20 mL/min and potassium is manageable
- At very advanced CKD (eGFR <15 or severe hyperkalemia), ACEi/ARB may need to be held while preparing for renal replacement therapy
- Add finerenone if still proteinuric on maximal RAAS blockade
- Add SGLT2 inhibitor if eGFR still >20 mL/min/1.73 m²
- Harrison's Principles of Internal Medicine, 22nd ed. (2025), pp. 2467+ (Pathogenesis, Natural History, Management, ACEi/ARB guidelines, Metformin)
- Robbins & Kumar Basic Pathology, p. 751 (Histological lesions - Kimmelstiel-Wilson nodule, diffuse sclerosis, arteriolosclerosis)
- Brenner and Rector's The Kidney (Epidemiology, Genetics, Pregnancy and DN)
- The Harriet Lane Handbook, 23rd ed. (Metformin eGFR contraindication)
Sarcoidosis Cirrhosis of Liver Diabetes Mellitus complications (DKA, Diabetic Kidney Disease) ARDS Pulmonary Embolism Cardiac Arrhythmias (AF, PSVT, Heart Block) COPD & Asthma Chronic Kidney Disease / Glomerulonephritis Hyponatremia Upper GI Bleeding
📚 Comprehensive Medical Study Guide
1. SARCOIDOSIS
Definition
Epidemiology
- Affects women > men (particularly women of childbearing age)
- African Americans > Whites in the USA (3-fold higher incidence)
- Peak incidence: 20-40 years; second peak in women >50
- Worldwide prevalence: Scandinavians, African Americans most affected
Etiology & Pathogenesis
- Unknown trigger (possibly mycobacterial or propionibacterial antigens in genetically susceptible individuals)
- Pathogenesis:
- Antigen presented by macrophages to CD4⁺ T helper cells (Th1 subtype)
- T cell activation → release of IL-2, IFN-γ, TNF-α
- Macrophage activation → epithelioid cell transformation
- Aggregation of epithelioid cells + multinucleated giant cells (Langhans type) → non-caseating granuloma
- Granulomas may resolve or progress to fibrosis (in ~20%)
- ACE (angiotensin-converting enzyme) is produced by epithelioid macrophages in granulomas → elevated serum ACE is a useful biomarker
Histology
- Non-caseating (non-necrotizing) epithelioid granuloma - pathognomonic
- Central zone: epithelioid histiocytes + multinucleated giant cells (Langhans or foreign body type)
- Peripheral rim: CD4⁺ T lymphocytes
- Schaumann bodies (laminated calcified concretions) and asteroid bodies (star-shaped inclusions) inside giant cells - characteristic but not pathognomonic
- No central caseation (distinguishes from TB)
Organs Involved
| Organ | Frequency | Features |
|---|---|---|
| Lungs | 90% | Bilateral hilar lymphadenopathy (BHL), interstitial infiltrates, restrictive PFTs |
| Lymph nodes | 75% | BHL on CXR; mediastinal, peripheral nodes |
| Skin | 25-35% | Erythema nodosum (acute), lupus pernio (chronic - alar rim; indicates upper airway involvement), maculopapular lesions |
| Eyes | 25% | Anterior uveitis (most common), posterior uveitis, conjunctival nodules, keratoconjunctivitis sicca |
| Liver | 60-70% (histological) | Usually asymptomatic; rarely cirrhosis |
| Heart | 5% clinically, 25% autopsy | Heart block (most common), arrhythmias, cardiomyopathy; major cause of sudden death in sarcoidosis |
| CNS | 5-15% | CN VII palsy (most common), CN II (optic nerve), hypothalamic-pituitary dysfunction, meningitis |
| Bones/joints | Lupus pernio + bone cysts (Phalangeal) | |
| Kidneys | 5-10% | Hypercalciuria/hypercalcemia (granulomas produce 1α-hydroxylase → ↑ active vitamin D → ↑ Ca absorption) |
| Parotid/salivary glands | Heerfordt syndrome: parotitis + uveitis + CN VII palsy + fever |
Staging (Chest X-Ray)
| Stage | CXR Finding | Spontaneous Resolution |
|---|---|---|
| 0 | Normal | - |
| I | Bilateral hilar lymphadenopathy (BHL) only | 60-80% |
| II | BHL + pulmonary infiltrates | 40-70% |
| III | Pulmonary infiltrates without BHL | 10-20% |
| IV | Pulmonary fibrosis | Irreversible |
Clinical Presentations
- Löfgren syndrome (acute benign): BHL + erythema nodosum + bilateral ankle periarthritis + fever. Excellent prognosis; resolves spontaneously in >90%.
- Heerfordt syndrome (uveoparotid fever): parotitis + uveitis + facial nerve palsy
- Chronic/insidious onset: progressive dyspnoea, dry cough, fatigue, weight loss
- Asymptomatic: incidental BHL on CXR in 30-50%
Investigations
- Serum ACE: elevated in 60-80% (non-specific; also elevated in TB, lymphoma, histoplasmosis)
- CXR / HRCT chest: BHL ± infiltrates; HRCT shows perilymphatic nodules, beading along bronchovascular bundles
- PFTs: Restrictive pattern (↓TLC, ↓VC, ↓DLCO); obstructive if endobronchial involvement
- Bronchoscopy + BAL: CD4:CD8 ratio >3.5 (normal ~1.8) in BAL fluid - highly suggestive
- Bronchoscopic biopsy (endobronchial or transbronchial): non-caseating granulomas
- Serum calcium: elevated (from ectopic 1,25-OH-vitamin D production by granulomas)
- 24-hr urine calcium: elevated → hypercalciuria → nephrolithiasis risk
- LFTs, renal function
- ECG: heart block, arrhythmias (Holter monitoring if cardiac sarcoidosis suspected)
- Ophthalmologic evaluation: slit-lamp exam
- Gallium-67 scan / FDG-PET: detects active granulomatous inflammation; "panda sign" (parotid + lacrimal gland uptake) + "lambda sign" (BHL)
Management
- Stage I, asymptomatic: Observe - spontaneous resolution likely
- Indications for systemic corticosteroids:
- Symptomatic pulmonary disease (Stage II-III)
- Cardiac, neurological, or ocular involvement
- Hypercalcemia
- Progressive disease
- Corticosteroid dosing: Prednisolone 20-40 mg/day for 6-12 months (or longer)
- Steroid-sparing agents: Methotrexate, azathioprine, hydroxychloroquine (for cutaneous/musculoskeletal disease)
- Anti-TNF agents: Infliximab for refractory disease
- Topical corticosteroids: For skin lesions (intralesional injection preferred)
- Cardiac sarcoidosis: ICD if high-risk for sudden cardiac death; pacemaker for complete heart block
- Prognosis: 2/3 patients have spontaneous remission within 3 years; ~10% develop progressive fibrotic disease
2. CIRRHOSIS OF THE LIVER
Definition
Etiology
| Cause | Notes |
|---|---|
| Alcohol (35-40%) | Most common in Western countries; threshold ~80 g/day for 10+ years |
| Chronic viral hepatitis - HCV (25%), HBV (15%) | Most common worldwide |
| NAFLD/NASH | Increasingly most common in developed countries |
| Wilson's disease | Copper accumulation; young patients |
| Hemochromatosis | Iron overload; "bronze diabetes" |
| Primary biliary cholangitis (PBC) | Anti-mitochondrial antibodies (AMA) |
| Primary sclerosing cholangitis (PSC) | Associated with IBD (UC) |
| Autoimmune hepatitis | Anti-smooth muscle antibodies (ASMA) |
| Budd-Chiari syndrome | Hepatic vein thrombosis |
| α1-antitrypsin deficiency | PAS-positive globules in hepatocytes |
| Cardiac cirrhosis | Right heart failure → chronic venous congestion |
Pathogenesis
- Chronic hepatic injury (any cause) → hepatocyte necrosis and inflammation
- Stellate cells (Ito cells) are the key effectors of fibrosis - activated by TGF-β, TNF-α, PDGF from injured hepatocytes and Kupffer cells
- Activated stellate cells transform into myofibroblasts → secrete collagen types I and III → fibrosis
- Fibrosis distorts liver architecture → impaired hepatocyte function + vascular distortion → portal hypertension
- Ongoing injury + impaired regeneration → end-stage cirrhosis
Portal Hypertension - Consequences
| Complication | Mechanism |
|---|---|
| Esophageal/gastric varices | Portosystemic collaterals (portal → azygous via submucosal esophageal veins) |
| Caput medusae | Portal → umbilical vein → superficial abdominal veins |
| Hemorrhoids (anorectal varices) | Portal → inferior rectal veins |
| Ascites | ↑ portal pressure + ↓ albumin (↓ oncotic pressure) + secondary hyperaldosteronism (RAAS activation from "underfill") → sodium/water retention |
| Splenomegaly → Hypersplenism | Congestion → pancytopenia (thrombocytopenia, leukopenia, anemia) |
Hepatic Dysfunction - Consequences
- Jaundice: impaired bilirubin conjugation and excretion
- Coagulopathy: reduced synthesis of factors II, V, VII, IX, X, fibrinogen
- Hypoalbuminemia: reduced albumin synthesis → edema, ascites
- Hepatic encephalopathy: impaired ammonia detoxification → cerebral dysfunction
- Hyperestrogenism: impaired estrogen metabolism → spider nevi, palmar erythema, gynecomastia, testicular atrophy, loss of axillary/pubic hair
- Hypoglycemia: reduced gluconeogenesis and glycogen storage
- Hepatorenal syndrome: functional renal failure (splanchnic vasodilation → renal vasoconstriction)
Clinical Features
- Spider nevi (>5 in upper body = significant), palmar erythema, leukonychia, finger clubbing, Dupuytren's contracture (alcohol)
- Parotid enlargement, gynecomastia, testicular atrophy, loss of body hair
- Jaundice, scleral icterus
- Liver: initially enlarged (fatty liver, hepatitis), eventually shrunken and firm in end-stage
- Ascites: shifting dullness, fluid thrill (in large ascites), caput medusae
- Splenomegaly
- Fetor hepaticus: sweet, musty odor from mercaptans (portosystemic shunting)
- Asterixis ("liver flap"): coarse tremor - sign of hepatic encephalopathy
Child-Pugh Score (Severity Assessment)
| Parameter | 1 | 2 | 3 |
|---|---|---|---|
| Bilirubin (mg/dL) | <2 | 2-3 | >3 |
| Albumin (g/dL) | >3.5 | 2.8-3.5 | <2.8 |
| PT prolongation (sec) | <4 | 4-6 | >6 |
| Ascites | None | Mild | Moderate-severe |
| Encephalopathy | None | Grade 1-2 | Grade 3-4 |
| Score | Class A (5-6) | Class B (7-9) | Class C (10-15) |
| 1-year survival | 100% | 80% | 45% |
Investigations
- LFTs: ↑AST, ↑ALT (AST:ALT ratio >2 suggests alcoholic liver disease), ↑ALP, ↑GGT (alcohol), ↑bilirubin
- Coagulation: ↑PT/INR (marker of synthetic function)
- Albumin: reduced (marker of chronic synthetic dysfunction)
- FBC: thrombocytopenia, anemia (multifactorial), leukopenia
- Urea/creatinine: hepatorenal syndrome
- Virology: HBsAg, anti-HCV, HCV RNA
- AFP (alpha-fetoprotein): screen for HCC
- Ultrasound abdomen: liver texture, portal vein diameter, ascites, focal lesions (HCC)
- Upper GI endoscopy: esophageal/gastric varices
- Liver biopsy: gold standard for staging fibrosis (Metavir F0-F4)
- Non-invasive fibrosis markers: FibroScan (transient elastography), FIB-4 score
Management
- Treat underlying cause (antiviral for HBV/HCV, abstinence from alcohol, weight loss for NAFLD)
- Nutritional support (protein 1.2-1.5 g/kg/day; avoid protein restriction)
- Vaccinations: Hepatitis A, B, pneumococcal, influenza
- Sodium restriction (2000 mg/88 mmol/day)
- Diuretics: Spironolactone 100 mg + Furosemide 40 mg daily (maintain 2.5:1 ratio); max: spironolactone 400 mg + furosemide 160 mg
- Large-volume paracentesis (LVP) for refractory ascites: 4-6 L with IV albumin 8 g per litre removed (prevents post-paracentesis circulatory dysfunction)
- TIPS (Transjugular Intrahepatic Portosystemic Shunt): for refractory ascites or refractory variceal bleeding
- Diagnosis: ascitic fluid PMN >250 cells/µL
- Treatment: IV cefotaxime 2g TDS × 5 days + IV albumin (1.5 g/kg day 1, 1 g/kg day 3)
- Prophylaxis (secondary): norfloxacin 400 mg BD; ciprofloxacin long-term
- Resuscitate; terlipressin (vasopressin analogue) or octreotide + IV antibiotics (norfloxacin)
- Urgent endoscopic variceal band ligation (EVL)
- Non-selective beta-blockers (propranolol, carvedilol) for primary and secondary prophylaxis
- TIPS for refractory bleeding
- Identify and treat precipitants (infection, GI bleed, constipation, sedatives, electrolyte disturbance)
- Lactulose (30 mL TDS, titrate to 2-3 soft stools/day): reduces ammonia production
- Rifaximin (550 mg BD): non-absorbable antibiotic; reduces recurrence
- Correct hypokalemia (worsens encephalopathy)
- Stop diuretics, NSAIDs, nephrotoxic drugs
- Type 1 HRS (acute): Terlipressin + albumin (1 g/kg/day)
- Type 2 HRS (chronic): TIPS; bridge to transplant
- Liver transplantation is definitive treatment
- 6-monthly ultrasound ± AFP in all cirrhotic patients
- Indications: Child-Pugh C, MELD ≥15, refractory complications, HCC within Milan criteria
3. DIABETES MELLITUS COMPLICATIONS
| Complication | Type | Key Features |
|---|---|---|
| DKA | Acute metabolic (mainly T1DM) | Glucose >250, pH <7.3, ketones - covered in detail above |
| HHS | Acute metabolic (T2DM) | Glucose >600, pH >7.3, no ketones, osmolality >320 |
| Hypoglycemia | Acute | BG <70 mg/dL; Rule of 15 |
| Retinopathy | Microvascular | Background → pre-proliferative → proliferative → blindness |
| Nephropathy | Microvascular | Microalbuminuria → proteinuria → CKD → ESKD - covered above |
| Neuropathy | Microvascular | Distal symmetric polyneuropathy, autonomic neuropathy |
| Macrovascular | Atherosclerosis | CAD, stroke, PAD - 2-4x higher mortality |
| Foot ulcer | Combined | Neuropathy + vasculopathy + infection |
4. ARDS (ACUTE RESPIRATORY DISTRESS SYNDROME)
Definition (Berlin 2012 Criteria)
| Severity | PaO₂/FiO₂ (P:F ratio) | PEEP |
|---|---|---|
| Mild | 200-300 mmHg | ≥5 cmH₂O |
| Moderate | 100-200 mmHg | ≥5 cmH₂O |
| Severe | <100 mmHg | ≥5 cmH₂O |
Common Causes
| Direct (Pulmonary) | Indirect (Extrapulmonary) |
|---|---|
| Pneumonia (most common) | Sepsis (most common overall - 40%) |
| Aspiration of gastric contents | Severe trauma/burns |
| Pulmonary contusion | Pancreatitis |
| Near-drowning | Multiple blood transfusions (TRALI) |
| Inhalation injury | DIC |
| Cardiopulmonary bypass |
Pathophysiology
- Injury to alveolar-capillary barrier (Type I pneumocytes + endothelium)
- Disruption of the endothelial tight junctions → protein-rich fluid floods alveoli ("non-cardiogenic pulmonary edema")
- Activation of neutrophils → release of proteases, ROS, cytokines (IL-1β, IL-6, IL-8, TNF-α) → further injury
- Loss of surfactant (Type II pneumocyte injury) → alveolar collapse (atelectasis) → reduced compliance
- Diffuse alveolar damage (DAD): hyaline membrane formation, alveolar flooding, hemorrhage
- V/Q mismatch + intrapulmonary shunt → refractory hypoxemia
- Resolution of edema; proliferation of Type II pneumocytes (attempt at repair)
- Fibroblast infiltration begins
- Clinical improvement in most patients
- In ~30% → progressive fibrosis replaces normal lung tissue
- Obliteration of alveoli and capillaries
- "Honeycombing" pattern on CT; fixed pulmonary fibrosis; chronic respiratory failure
Clinical Features
- Acute onset (within 1 week of precipitant)
- Severe dyspnoea, tachypnoea, use of accessory muscles
- Refractory hypoxemia (PaO₂ fails to improve with supplemental O₂ alone)
- Diffuse bilateral crackles
- Cyanosis in severe disease
- CXR: bilateral diffuse alveolar opacities ("white-out lungs") without cardiomegaly
- HRCT: diffuse bilateral ground-glass opacities, consolidation, dependent atelectasis ("crazy paving")
Investigations
- ABG: hypoxemia (↓PaO₂), initial respiratory alkalosis → may develop metabolic acidosis
- P:F ratio: key measure of severity
- CXR/CT chest: bilateral opacities
- Echocardiography: to exclude cardiogenic pulmonary edema (PCWP <18 mmHg in ARDS)
- BNP/proBNP: low in ARDS (high in cardiogenic)
- Identify and investigate the underlying cause: blood/sputum cultures, procalcitonin, pancreatitis markers, etc.
Management
- Low tidal volume ventilation: Vt = 6 mL/kg ideal body weight (ARDSnet protocol)
- Plateau pressure: <30 cmH₂O
- PEEP: titrated to improve oxygenation (typically 5-15 cmH₂O)
- Target SpO₂: 88-95%; accept permissive hypercapnia (PaCO₂ up to 55-60 mmHg)
- Prone positioning: ≥16 hours/day in moderate-severe ARDS (P:F <150) → reduces mortality (~16% absolute reduction; PROSEVA trial)
- Conservative fluid strategy (once hemodynamically stable) reduces duration of ventilation
- Target euvolemia; avoid excessive fluid resuscitation after initial resuscitation
- 48 hours of cisatracurium infusion in early severe ARDS (P:F <150) - reduces ventilator dyssynchrony and inflammation (ACURASYS trial, though ROSE trial showed less benefit)
- Controversial; may reduce duration of ventilation in moderate-severe ARDS if given within 14 days of onset; dexamethasone 20 mg/day × 5 days, then 10 mg/day × 5 days
- Antibiotics for pneumonia/sepsis; treat pancreatitis, remove toxic exposures
- DVT prophylaxis (LMWH + compression stockings)
- Stress ulcer prophylaxis (PPI or H2RA)
- Early enteral nutrition
- Ventilator-associated pneumonia (VAP) prevention bundles
- Glucose control (target 140-180 mg/dL)
- Avoid unnecessary sedation; daily sedation holds
- For refractory ARDS (P:F <80 despite optimal mechanical ventilation); referral to specialist ECMO center (CESAR/EOLIA trials)
5. PULMONARY EMBOLISM (PE)
Definition
Risk Factors - Virchow's Triad
| Component | Examples |
|---|---|
| Stasis | Immobility (prolonged travel, surgery, ICU), cardiac failure, obesity |
| Endothelial injury | Surgery, trauma, prior DVT, indwelling catheters |
| Hypercoagulability | Inherited thrombophilia (Factor V Leiden, Prothrombin mutation, Protein C/S deficiency, Antithrombin III deficiency), OCP/HRT, pregnancy, malignancy, antiphospholipid syndrome, nephrotic syndrome |
Pathophysiology
- Thrombus detaches from DVT (usually ilio-femoral veins) → travels to pulmonary circulation
- Hemodynamic consequences: obstruction of pulmonary arterial bed → ↑ pulmonary vascular resistance (PVR) → ↑ right ventricular (RV) afterload → RV strain → RV dilation and failure → leftward septal shift → ↓ LV filling → ↓ CO → cardiogenic shock (in massive PE)
- Respiratory consequences:
- Dead space (obstructed segments) + low V/Q zones (redistribution of blood to non-obstructed areas) → hypoxemia (see Murray & Nadel above)
- Loss of surfactant → atelectasis → shunting (in large PE after 24-48 hours)
- Reflex bronchoconstriction (serotonin, histamine from platelets in embolus)
- Hyperventilation → hypocapnia (hypocarbia) - most common ABG finding
- Rising PCO₂ during acute PE = sign of massive obstruction/inability to compensate → ominous
Classification
| Type | Definition |
|---|---|
| Massive PE | Hemodynamic instability (SBP <90 mmHg, cardiac arrest); RV failure |
| Submassive PE | Hemodynamically stable but evidence of RV dysfunction/myocardial injury (↑troponin, ↑BNP, RV dilation on echo/CT) |
| Low-risk PE | Hemodynamically stable, no RV dysfunction, ↓troponin/BNP |
Clinical Features
- Dyspnoea (most common, 80%)
- Pleuritic chest pain (40-70%) - infarction of lung near pleura
- Haemoptysis (10-20%) - pulmonary infarction
- Tachycardia (most common sign)
- Tachypnoea
- Syncope (in massive PE)
- Signs of DVT: calf pain, swelling, redness in lower limb (only 25% have concurrent symptomatic DVT)
- Massive PE: hypotension, raised JVP, loud P2, RV gallop, parasternal heave
Investigations
- D-dimer: sensitive but not specific; negative D-dimer (<500 ng/mL) in low-probability PE effectively excludes it
- ABG: hypoxemia, hypocapnia (respiratory alkalosis), ↑A-a gradient; hypocarbia typical
- CXR: usually normal; may show Hampton's hump (peripheral wedge-shaped opacity), Westermark sign (oligaemia of lung zone), atelectasis, pleural effusion
- ECG: sinus tachycardia (most common); classic S1Q3T3 pattern (S in lead I, Q wave and inverted T in lead III) - seen in only 20%; RV strain (T-wave inversions in V1-V4); RBBB
- CT Pulmonary Angiography (CTPA): gold standard investigation; shows filling defects in pulmonary arteries; also assesses RV:LV ratio
- Ventilation-Perfusion (V/Q) scan: alternative if CTPA contraindicated (renal failure, contrast allergy, pregnancy); shows perfusion defects without matched ventilation defects
- Echocardiography: RV dilation, McConnell's sign (RV free wall hypokinesis with apical sparing), septal flattening (D-sign); useful in hemodynamically unstable patients
- Troponin, BNP/proBNP: elevated in submassive/massive PE → prognostic markers
- WELLS score / Geneva score: pre-test clinical probability; guides investigation pathway
- Lower limb Doppler ultrasound: detects DVT (if positive, no further PE investigation needed before anticoagulation)
Management
- Immediate systemic thrombolysis: Alteplase 100 mg IV over 2 hours
- If thrombolysis contraindicated: surgical embolectomy or catheter-directed therapy
- Anticoagulation immediately after thrombolysis
- Anticoagulation (see below) - standard treatment
- Consider thrombolysis in deteriorating patients (individualize)
- Close monitoring; HDU/ICU setting
- Anticoagulation alone
- Consider outpatient treatment if low PESI score (PESI class I-II)
- DOACs (preferred): Rivaroxaban 15 mg BD × 3 weeks, then 20 mg OD; OR Apixaban 10 mg BD × 7 days, then 5 mg BD
- LMWH + warfarin: Enoxaparin bridge with warfarin (INR target 2-3); overlap at least 5 days until INR ≥2 for 24 hours
- Duration:
- Provoked (reversible cause): 3 months
- Unprovoked: ≥3-6 months; consider indefinite
- Active cancer: LMWH or DOAC (edoxaban, rivaroxaban) indefinitely
- Recurrent PE or antiphospholipid syndrome: indefinite
- VTE prophylaxis in hospitalized patients (LMWH + mechanical compression stockings)
- Early ambulation post-surgery
- Inferior vena cava (IVC) filter: if anticoagulation contraindicated (active bleeding)
6. CARDIAC ARRHYTHMIAS
A. Atrial Fibrillation (AF)
Definition
Classification
- Paroxysmal: episodes <7 days, self-terminating
- Persistent: >7 days, requires cardioversion
- Long-standing persistent: >12 months
- Permanent: ongoing; rhythm control abandoned
Causes (PIRATES mnemonic)
- Pericarditis/Pulmonary embolism/Pneumonia
- Ischaemic heart disease (MI)
- Rheumatic heart disease (mitral valve disease)
- Alcohol ("holiday heart"), Anemia
- Thyrotoxicosis (one of the most important reversible causes)
- Essential hypertension (most common associated condition), Electrolyte disturbance
- Sepsis, Surgery (particularly cardiac), Structural heart disease (cardiomyopathy, heart failure)
Pathophysiology
- Multiple wavelet reentry within the atria (substrate: fibrosis, inflammation)
- Focal triggers: pulmonary vein orifices (ectopic foci) in paroxysmal AF
- Rapid irregular ventricular response (120-180 bpm) → irregular palpitations, reduced diastolic filling → reduced CO
- Stasis of blood in left atrial appendage → thrombus formation → embolism → stroke (main complication)
Clinical Features
- Palpitations, dyspnoea, fatigue, reduced exercise tolerance
- Irregular pulse; deficit between apical and radial pulse
- Hypotension if rapid ventricular rate
- Signs of heart failure (if pre-existing dysfunction)
- ECG: Absent P waves; irregularly irregular RR intervals; fibrillatory baseline; narrow QRS (unless aberrant conduction or accessory pathway)
Complications
- Stroke / TIA (5× increased risk without anticoagulation)
- Heart failure / tachycardiomyopathy
- Hemodynamic compromise
Assessment
- ECG (12-lead); Holter monitor (for paroxysmal AF)
- Echo: assess LV function, valvular disease, LA size, thrombus in LAA (TEE more sensitive)
- TFTs (thyroid), FBC, electrolytes, renal function
- CHA₂DS₂-VASc score: assess stroke risk
CHA₂DS₂-VASc Score (Stroke Risk)
| Factor | Points |
|---|---|
| Congestive heart failure | 1 |
| Hypertension | 1 |
| Age ≥75 | 2 |
| Diabetes mellitus | 1 |
| Stroke/TIA history | 2 |
| Vascular disease (MI, PAD) | 1 |
| Age 65-74 | 1 |
| Sex category (female) | 1 |
- Score ≥2 (men) or ≥3 (women): anticoagulate
- Score 1 (men): consider anticoagulation
Management
- Target resting HR <110 bpm (lenient) or <80 bpm (strict)
- Drugs: Beta-blockers (metoprolol, bisoprolol), Digoxin (particularly in heart failure/sedentary), Diltiazem/Verapamil (calcium channel blockers - avoid in HFrEF)
- DC cardioversion: synchronized 200 J biphasic (anticoagulate for ≥3 weeks before if AF >48 hours, or TEE to exclude LAA thrombus)
- Pharmacological cardioversion: Flecainide or propafenone (no structural disease); Amiodarone (structural heart disease)
- Catheter ablation (pulmonary vein isolation - PVI): for paroxysmal AF refractory to drugs; increasingly first-line in young patients
- DOACs preferred: Rivaroxaban, Apixaban, Dabigatran, Edoxaban
- Warfarin: INR 2-3; used in valvular AF (especially mechanical valves, moderate-severe mitral stenosis)
- HAS-BLED score: assess bleeding risk before anticoagulation
B. Paroxysmal Supraventricular Tachycardia (PSVT)
Definition
Mechanism
- AVNRT: Dual AV nodal pathways (fast/slow) → reentry circuit confined to AV node
- AVRT: Accessory pathway (Bundle of Kent in WPW) bypasses the AV node → reentry loop between AV node (antegrade) and accessory pathway (retrograde)
Clinical Features
- Sudden onset palpitations ("heart racing"; "flip-flop in chest")
- Neck pulsations (AV dissociation/simultaneous atrial and ventricular contraction)
- Dyspnoea, light-headedness, syncope (rare)
- Sudden termination
- ECG during tachycardia: Narrow QRS regular tachycardia (150-250 bpm); P waves may be hidden within QRS (AVNRT) or shortly after QRS (AVRT); no delta waves during PSVT (unless anterograde accessory pathway conduction)
- WPW pre-excitation on resting ECG: Short PR interval (<120 ms) + delta wave (slurred QRS upstroke) + wide QRS
Management
- Vagal maneuvers first: Valsalva (supine, 40 mmHg sustained) → "Modified Valsalva" (legs elevated after), carotid sinus massage (in young patients)
- IV Adenosine (6 mg rapid IV bolus; if no response: 12 mg, then 12 mg again): blocks AV node conduction; drug of choice for acute termination; brief asystole is expected and alarming but transient
- Adenosine contraindicated in: severe asthma, WPW with AF (can cause VF), heart transplant recipients
- IV Verapamil (5-10 mg slow IV): alternative if adenosine fails
- DC cardioversion (synchronized): if hemodynamically unstable
- Catheter ablation (RFA - Radiofrequency Ablation): highly effective (>95% cure rate); first-line for symptomatic/recurrent PSVT
- Beta-blockers or flecainide: medical prophylaxis if patient declines ablation
C. Heart Block
First-Degree Heart Block
- Prolonged PR interval >200 ms (>5 small squares) on ECG
- Every P wave followed by QRS; 1:1 conduction
- Usually benign; no treatment required
- Causes: increased vagal tone (athletes), inferior MI, digoxin, beta-blockers, calcium channel blockers
Second-Degree Heart Block
- Progressive PR prolongation until a P wave is not conducted (QRS dropped)
- Pattern repeats in cycles (e.g., 3:2, 4:3)
- Usually benign (AV nodal level); responds to atropine; rarely needs pacing
- Causes: Inferior MI (RCA occlusion), digoxin toxicity
- Fixed PR interval; sudden non-conduction of P wave without preceding PR prolongation
- Location: below AV node (His-Purkinje system) → unpredictable complete block → Stokes-Adams attacks (syncope)
- Serious - high risk of progression to complete heart block
- Requires permanent pacing
Third-Degree (Complete) Heart Block
- Complete dissociation between atrial and ventricular activity
- ECG: Regular P waves at own rate (60-100/min) + Regular QRS at escape rate (idioventricular 20-40/min); no relationship between P and QRS
- Clinical: Cannon A waves (atria contracting against closed AV valves), variable S1 intensity, Stokes-Adams attacks (sudden loss of consciousness due to asystole/VT/VF)
- Causes: Inferior MI (usually transient), anterior MI (permanent), Lyme disease, sarcoidosis, complete cardiac sarcoidosis, post-surgical, degenerative (Lev's and Lenègre's disease), congenital
- Management: Emergency - temporary pacing (transcutaneous or transvenous); permanent pacemaker (required in most cases)
7. COPD & ASTHMA
COPD (Chronic Obstructive Pulmonary Disease)
Definition (GOLD 2023)
COPD vs Asthma Key Differences
| Feature | COPD | Asthma |
|---|---|---|
| Age of onset | Usually >40 years | Usually <25 years |
| Smoking history | Almost universal (90%+) | Not required |
| Symptoms | Persistent, progressive | Episodic, variable |
| Airflow obstruction | Fixed / incompletely reversible | Fully reversible |
| FEV₁ reversibility | <12% or <200 mL | ≥12% AND ≥200 mL |
| Diurnal variation | Absent | Present (>20% PEFR variation) |
| Atopy | Uncommon | Common |
| Eosinophilia | Less prominent | Common |
| Predominant cells | Neutrophils, CD8⁺ T cells, macrophages | Eosinophils, mast cells, CD4⁺ T cells |
| CXR | Hyperinflation, flat diaphragms, bullae | Usually normal |
| DLCO | Reduced (emphysema destroys alveolar surface) | Normal or increased |
Types of COPD
- Chronic Bronchitis: Productive cough for ≥3 months in each of ≥2 consecutive years ("blue bloaters" - hypercapnic, cyanotic, edematous due to cor pulmonale)
- Emphysema: Destruction of alveolar walls with air space enlargement ("pink puffers" - hyperventilate to maintain oxygenation, thin with pursed-lip breathing)
- Most patients have both components
GOLD Staging (post-bronchodilator FEV₁% predicted)
| GOLD Grade | FEV₁ % Predicted | Severity |
|---|---|---|
| 1 | ≥80% | Mild |
| 2 | 50-79% | Moderate |
| 3 | 30-49% | Severe |
| 4 | <30% | Very severe |
Pathogenesis
- Cigarette smoke → activation of innate and adaptive immunity → neutrophils + macrophages + CD8⁺ T cells in airways
- Oxidative stress → protease/antiprotease imbalance: ↑ MMP (matrix metalloproteinase), ↓ α1-antitrypsin → emphysema (alveolar wall destruction)
- Chronic airway inflammation → goblet cell hyperplasia, smooth muscle hypertrophy, subepithelial fibrosis → chronic bronchitis (large airways) + small airway disease
- Air trapping → dynamic hyperinflation → worsening dyspnoea with exertion
Management
- Smoking cessation (most effective intervention to slow progression)
- Vaccinations: annual influenza, pneumococcal (PCV13 + PPSV23), COVID-19
- Pulmonary rehabilitation (for MRC dyspnoea grade ≥3)
- Supplemental oxygen if PaO₂ <55 mmHg (or <60 mmHg with cor pulmonale/polycythemia): improves survival
- SABA (salbutamol) as reliever
- LAMA (tiotropium, umeclidinium): preferred long-acting bronchodilator in COPD
- LABA (salmeterol, formoterol, indacaterol): alternative or add-on
- LAMA + LABA dual bronchodilation: standard for symptomatic/moderate-severe COPD
- ICS indicated when: ≥2 exacerbations/year OR 1 hospitalization + blood eosinophils ≥300 cells/µL
- Triple therapy (LAMA + LABA + ICS): for high exacerbation risk
- ICS alone should NOT be used in COPD (unlike asthma)
- Roflumilast (PDE4 inhibitor): reduces exacerbations in severe COPD with chronic bronchitis phenotype
- Azithromycin (long-term, low dose): reduces exacerbations in selected patients (ex-smokers)
- N-acetylcysteine: mucolytic; some evidence for exacerbation reduction
- Increased dyspnoea, sputum volume, sputum purulence (Anthonisen criteria)
- Triggers: viral URTI (rhinovirus), bacterial (H. influenzae, S. pneumoniae, M. catarrhalis), air pollution
- Management:
- Controlled oxygen: target SpO₂ 88-92% (risk of hypercapnic respiratory failure in COPD - don't over-oxygenate)
- Bronchodilators: nebulized salbutamol + ipratropium (SAMA + SABA)
- Systemic corticosteroids: Prednisolone 30-40 mg/day × 5 days → faster recovery, reduced hospital stay
- Antibiotics: if purulent sputum, CRP >40 mg/L, or hospitalization required (amoxicillin, doxycycline, or azithromycin; co-amoxiclav if severe)
- NIV (Non-Invasive Ventilation): CPAP/BiPAP for respiratory acidosis (pH <7.35, PaCO₂ >6.0 kPa); reduces intubation and mortality
- ICU/invasive ventilation: if NIV fails or contraindicated
8. CHRONIC KIDNEY DISEASE (CKD) / GLOMERULONEPHRITIS
Chronic Kidney Disease (CKD)
Definition (KDIGO 2012)
Staging (GFR-based)
| Stage | GFR (mL/min/1.73 m²) | Description |
|---|---|---|
| G1 | ≥90 | Normal/high (with markers of damage) |
| G2 | 60-89 | Mildly decreased |
| G3a | 45-59 | Mildly-moderately decreased |
| G3b | 30-44 | Moderately-severely decreased |
| G4 | 15-29 | Severely decreased |
| G5 | <15 | Kidney failure (ESKD) |
Common Causes
- Diabetic nephropathy (35-40%) - most common in developed world
- Hypertensive nephrosclerosis (25%)
- Glomerulonephritis (15%)
- Polycystic kidney disease (5%)
- Chronic tubulointerstitial nephritis (analgesic nephropathy, reflux nephropathy)
- Renovascular disease
Complications of CKD
| System | Complication | Mechanism |
|---|---|---|
| Cardiovascular | Accelerated atherosclerosis, LVH, heart failure, arrhythmias | Hypertension, fluid overload, anemia, uremic toxins, dyslipidemia |
| Hematological | Normochromic normocytic anemia | ↓ EPO production by peritubular cells (see DKD section) |
| Metabolic | Hyperkalemia | ↓ Urinary K⁺ excretion; acidosis drives K⁺ out of cells |
| Acid-base | Metabolic acidosis | ↓ NH₃ synthesis for H⁺ excretion; ↓ HCO₃ reabsorption |
| Bone/mineral | CKD-MBD (Mineral-Bone Disease) | ↓ 1,25-OH vitamin D → ↓ Ca absorption → ↑ PTH (secondary hyperparathyroidism) → bone resorption (osteitis fibrosa cystica); hyperphosphatemia |
| Neurological | Uremic encephalopathy, peripheral neuropathy, restless legs | Uremic toxin accumulation |
| Dermatological | Pruritus, pallor, "uremic frost" | Uremic toxin deposition; anemia |
| GI | Nausea, vomiting, uremic pericarditis, GI bleeding | Uremia; platelet dysfunction |
| Immune | Increased infection risk | Neutrophil dysfunction, reduced opsonization |
| Fluid | Edema, hypertension | Salt and water retention |
Management
- Treat underlying cause
- Blood pressure control: target <130/80 mmHg; ACEi/ARB first-line (reduces proteinuria and GFR decline)
- Glycemic control in diabetics: HbA1c <7%
- SGLT2 inhibitors: now standard of care for CKD with T2DM (and increasingly for non-diabetic proteinuric CKD)
- Proteinuria reduction: ACEi/ARB; target UACR reduction >30%
- Anemia: Erythropoiesis-stimulating agents (ESA) - EPO/darbepoetin; iron supplementation; Hb target 10-12 g/dL
- CKD-MBD: Phosphate binders (calcium carbonate, sevelamer); active vitamin D (calcitriol); cinacalcet for secondary hyperparathyroidism
- Hyperkalemia management: Dietary restriction, patiromer or sodium zirconium cyclosilicate (novel K⁺ binders)
- Acidosis: Sodium bicarbonate supplementation (target serum HCO₃ >22 mEq/L)
- Renal replacement therapy: Hemodialysis, peritoneal dialysis, or renal transplantation when GFR <10-15 mL/min with uremic symptoms
Glomerulonephritis (GN)
Classification
| Type | Predominant Feature | Key Conditions |
|---|---|---|
| Nephritic syndrome | Hematuria, hypertension, oliguria, mild proteinuria (<3.5 g/day), azotemia | IgA nephropathy, post-streptococcal GN, RPGN, Goodpasture's, lupus nephritis |
| Nephrotic syndrome | Proteinuria >3.5 g/day, hypoalbuminemia <3 g/dL, edema, hyperlipidemia, lipiduria | Minimal change disease (children), FSGS, membranous nephropathy, diabetic nephropathy |
Common Glomerulonephritides
| Disease | Pathology | Typical Presentation | IF/EM Findings |
|---|---|---|---|
| IgA Nephropathy (Berger's) | Mesangial IgA deposits | Young adult; gross hematuria 1-3 days after URTI ("synpharyngitic hematuria") | IgA mesangial deposits on IF |
| Post-streptococcal GN | Immune complex | Child, 2-3 weeks after strep throat; nephritic syndrome; low C3 | "Humps" (subepithelial deposits) on EM; granular IgG + C3 on IF |
| Minimal Change Disease | Podocyte injury; no LM changes | Children: nephrotic syndrome; responds to steroids | Foot process effacement on EM; negative IF |
| Membranous Nephropathy | Subepithelial immune deposits; GBM thickening | Adults: nephrotic syndrome; anti-PLA2R antibodies; risk of thrombosis (renal vein) | "Spike and dome" on EM; granular IgG on IF |
| FSGS | Segmental sclerosis of glomeruli | Nephrotic syndrome; adults/AA; poor prognosis; HIV-associated (collapsing variant) | Focal segmental sclerosis on LM; foot process effacement on EM |
| Crescentic/RPGN | Crescent formation (cellular → fibrous) | Rapidly progressive renal failure over days-weeks; hematuria | Anti-GBM (Goodpasture's) / Immune complex / Pauci-immune (ANCA-positive: GPA, MPA) |
| Lupus Nephritis (WHO/ISN Class I-VI) | Immune complex; "full house" IF | Young female; SLE; proteinuria, hematuria, hypertension | IgG, IgM, IgA, C3, C1q all positive on IF ("full house") |
| Membranoproliferative GN | Mesangial proliferation + GBM thickening | Mixed nephritic-nephrotic; low C3 | "Tram-track" (double contour) on LM; EM type I-III |
Anti-GBM Disease (Goodpasture's Syndrome)
- Antibodies against type IV collagen in GBM and alveolar basement membrane
- Crescentic GN + pulmonary hemorrhage (hemoptysis)
- Linear IgG along GBM on immunofluorescence (pathognomonic)
- Treatment: Plasmapheresis + cyclophosphamide + corticosteroids
9. HYPONATREMIA
Definition
Classification by Osmolality
| Type | Serum Osmolality | Cause | Na⁺ Level Explanation |
|---|---|---|---|
| Hypertonic hyponatremia | >295 mOsmol/kg | Hyperglycemia, mannitol | Osmotic shift of water from ICF to ECF dilutes Na⁺ |
| Isotonic/pseudohyponatremia | 280-295 mOsmol/kg | Severe hyperlipidemia, hyperproteinemia | Lab artifact; water content of plasma falsely low |
| Hypotonic hyponatremia | <280 mOsmol/kg | Most clinical hyponatremia | True deficiency of Na⁺ relative to water |
Classification of Hypotonic Hyponatremia by Volume Status
1. Hypovolemic Hyponatremia (↓ECV + ↓Na⁺)
- Total body sodium LOW; water low but sodium even lower
- Renal losses (urine Na⁺ >20 mEq/L): diuretics (thiazides most common), salt-wasting nephropathy, adrenal insufficiency (↓aldosterone), cerebral salt wasting
- Extra-renal losses (urine Na⁺ <20 mEq/L): vomiting, diarrhea, sweating, burns, GI fistulae
- Treatment: IV 0.9% Normal saline (restores volume)
2. Euvolemic Hyponatremia (Normal ECV + ↑Total body water)
- Total body sodium NORMAL; water excess
- SIADH (most common cause of euvolemic hyponatremia)
- Hypothyroidism (↓cardiac output → ADH release)
- Adrenal insufficiency (cortisol normally inhibits ADH; cortisol deficiency → ↑ADH)
- Primary polydipsia: excessive water intake overwhelms renal diluting capacity
- Treatment: Fluid restriction; treat underlying cause; hypertonic saline in severe symptomatic cases
3. Hypervolemic Hyponatremia (↑ECV + ↑Na⁺ but ↑↑water)
- Total body sodium HIGH; water even higher
- Heart failure → ↓effective arterial volume → RAAS + ADH activation → water retention
- Cirrhosis → splanchnic vasodilation → ↓effective arterial volume → RAAS + ADH → water retention
- Nephrotic syndrome → ↓oncotic pressure → edema → ↓effective arterial volume
- Renal failure → inability to excrete free water
- Urine Na⁺ <20 mEq/L (RAAS activated)
- Treatment: Fluid restriction; treat underlying cause; diuretics; vaptans (tolvaptan) for refractory SIADH/cirrhosis
SIADH - Diagnostic Criteria (Schwartz-Bartter)
- Serum Na⁺ <135 mEq/L
- Serum osmolality <280 mOsmol/kg
- Urine osmolality >100 mOsmol/kg (inappropriately concentrated)
- Urine Na⁺ >40 mEq/L (kidneys retain ability to excrete Na⁺)
- No edema; euvolemic
- Normal adrenal and thyroid function
- No diuretic use
- CNS: meningitis, encephalitis, SAH, stroke, brain tumors, head trauma
- Lungs: pneumonia, TB, lung abscess, small cell lung carcinoma (ectopic ADH)
- Drugs: carbamazepine, SSRIs, TCAs, vincristine, cyclophosphamide, NSAIDs, chlorpropamide
- Other: postoperative, pain, nausea, HIV
Symptoms
- Serum Na⁺ >125: usually asymptomatic or mild (nausea, malaise, headache)
- Na⁺ 115-125: headache, confusion, disorientation, lethargy
- Na⁺ <115 or rapid fall: seizures, coma, brain herniation, death
Treatment
- Acute (<48 hours): can correct faster; significant brain edema risk
- Chronic (>48 hours or unknown): MUST correct slowly - rapid correction causes osmotic demyelination syndrome (ODS) - formerly called central pontine myelinolysis
- Chronic hyponatremia: No more than 8-10 mEq/L per 24 hours (or 12 mEq/L max)
- Severe neurological symptoms (seizures, coma): 3% hypertonic saline as bolus or infusion to raise Na⁺ by 4-6 mEq/L → resolves symptoms; then slow
| Cause | Treatment |
|---|---|
| Hypovolemic | 0.9% NS to restore volume |
| SIADH | Fluid restriction (500 mL less than urine output); tolvaptan (V2 receptor antagonist); demeclocycline |
| Heart failure | Diuretics + treat underlying HF |
| Cirrhosis | Fluid restriction; tolvaptan; treat underlying cirrhosis |
| Hypothyroidism | Thyroid hormone replacement |
| Adrenal insufficiency | Hydrocortisone |
| Hypervolemia (nephrotic) | Diuretics + treat underlying disease |
- Complication of overly rapid correction of chronic hyponatremia
- Demyelination of central pontine fibers + extrapontine areas
- Presents 2-6 days after correction: dysarthria, dysphagia, paraplegia/quadriplegia, "locked-in syndrome," coma
- MRI: hyperintensity in pons (trident sign)
- Largely irreversible; prevention is key
10. UPPER GASTROINTESTINAL BLEEDING
Definition
Presentation
- Hematemesis: vomiting of fresh blood (active bleeding) or "coffee-ground" material (old/altered blood)
- Melena: black, tarry, foul-smelling stool (hemoglobin degraded to hematin by gut bacteria); requires ≥50-100 mL blood in upper GI tract
- Hematochezia: bright red blood per rectum (usually lower GI source, but can be upper GI with rapid, massive bleeding ≥1000 mL)
Causes (Goldman-Cecil data + prevalence)
| Cause | % of Cases |
|---|---|
| Peptic ulcer disease (gastric/duodenal) | 38-40% |
| Esophageal/gastric varices (portal hypertension) | 16% |
| Erosive esophagitis / gastritis / duodenitis | 13% |
| Upper GI tumors (gastric cancer, GIST) | 7% |
| Angiodysplasia / vascular ectasia | 6% |
| Mallory-Weiss tear (longitudinal mucosal tear at gastroesophageal junction after retching/vomiting) | 4% |
| Dieulafoy lesion (abnormally large submucosal artery) | 2% |
| No cause found | 8% |
Risk Stratification Scores
- Glasgow-Blatchford Score (GBS): Before endoscopy; predicts need for intervention; score ≥1 = high risk; score 0 = safe for outpatient management
- Rockall Score: After endoscopy; predicts re-bleeding and mortality risk
Initial Assessment and Resuscitation
- Airway protection: Intubate if massive bleeding, altered consciousness (aspiration risk)
- 2 large-bore IV cannulae (≥16 gauge); send FBC, coagulation, U&E, LFTs, crossmatch (4-6 units)
- Fluid resuscitation: IV 0.9% NS or crystalloid; blood products if hemodynamically unstable
- Blood transfusion: Trigger Hb <7 g/dL (restrictive strategy - TRIGGER trial); Hb <8 g/dL in cardiovascular disease
- Reverse coagulopathy: Vitamin K if on warfarin; FFP + platelet transfusion if coagulopathic
- Monitor: HR, BP, urine output (catheter), CVP if needed
Management of Non-Variceal Upper GI Bleeding (Peptic Ulcer)
- IV PPI (Proton Pump Inhibitor): High-dose PPI infusion (esomeprazole/omeprazole 80 mg IV bolus, then 8 mg/hour for 72 hours post-endoscopy) - raises gastric pH > 6, stabilizes clot
- Start PPI before endoscopy; continue after endoscopic therapy
- Stop NSAIDs, aspirin (unless antithrombotic for secondary prevention - discuss with cardiologist)
- Timing: Within 24 hours of presentation (within 12 hours if hemodynamically unstable and resuscitated)
- Indications for intervention (Forrest classification):
| Forrest Class | Endoscopic Appearance | Re-bleed Risk | Treatment |
|---|---|---|---|
| Ia | Spurting arterial hemorrhage | 90% | Endoscopic therapy |
| Ib | Oozing hemorrhage | 50% | Endoscopic therapy |
| IIa | Visible vessel | 50% | Endoscopic therapy |
| IIb | Adherent clot | 25-30% | Endoscopic therapy (remove clot) |
| IIc | Flat pigmented spot | 7-10% | PPI alone |
| III | Clean base | <5% | PPI alone; early discharge |
- Endoscopic hemostatic techniques: Adrenaline injection (1:10,000) + thermal coagulation (bipolar/argon plasma coagulation) or mechanical clips; "dual therapy" preferred
- Test all peptic ulcer bleeds for H. pylori (rapid urease test on biopsy, or stool antigen/urea breath test after 4 weeks)
- Eradication with triple therapy (PPI + amoxicillin + clarithromycin × 7-14 days) dramatically reduces re-bleeding
- Repeat endoscopy with attempt at endoscopic hemostasis
- Interventional radiology (IR) embolization: if endoscopic treatment fails
- Surgery: if IR fails; Billroth I/II gastrectomy or oversewing of vessel
Management of Variceal Upper GI Bleeding
- Terlipressin (first-line): 2 mg IV 4-hourly × 24 hours, then 1 mg × 72 hours total; reduces portal pressure (vasopressin V1 agonist → splanchnic vasoconstriction)
- Alternative: Octreotide/Somatostatin - reduces splanchnic blood flow
- Norfloxacin 400 mg BD or ceftriaxone 1 g/day IV × 5-7 days
- Reduces SBP, reduces re-bleeding, improves survival (all cirrhotic patients with GI bleed)
- Endoscopic variceal band ligation (EVL): preferred over sclerotherapy for esophageal varices
- N-butyl cyanoacrylate injection ("glue"): for gastric varices (EVL less effective here)
- Temporary measure only (maximum 24 hours) - bridge to definitive treatment when endoscopy fails
- Associated with serious complications (aspiration, esophageal necrosis/rupture)
- For variceal bleeding refractory to endoscopic + pharmacological treatment
- Also used as "early TIPS" within 72 hours of index bleed in high-risk patients (Child-Pugh B with active bleeding, or Child-Pugh C)
- Connects portal vein to hepatic vein via liver stent → decompresses portal system
- Complications: hepatic encephalopathy (increased), stent stenosis
- Non-selective beta-blockers: Propranolol (start 40 mg BD, increase to max tolerated) or Carvedilol
- Repeated EVL sessions until variceal obliteration
- Both together (NSBB + EVL) more effective than either alone
- Goldman-Cecil Medicine (Sarcoidosis, Upper GI Bleeding, Atrial Fibrillation)
- Brenner & Rector's The Kidney (CKD, Cirrhosis/Ascites, Hyponatremia)
- Murray & Nadel's Textbook of Respiratory Medicine (COPD, Asthma, Pulmonary Embolism, ARDS)
- Robbins & Kumar Basic Pathology (Sarcoidosis pathology, Glomerulonephritis, Diabetic Nephropathy)
- Harrison's Principles of Internal Medicine, 22nd Ed. (2025) (Diabetic Nephropathy, CKD management)
- Braunwald's Heart Disease (Cardiac Arrhythmias - AF, PSVT, Heart Block)
- Textbook of Family Medicine, 9th Ed. (COPD, Sarcoidosis overview)
- Fishman's Pulmonary Diseases and Disorders (ARDS)
Cirrhosis of Liver
"liver cirrhosis"[MeSH Terms] AND "complications"[MeSH Terms]
CIRRHOSIS OF THE LIVER
A Comprehensive Textbook-Level Study Guide
1. DEFINITION
- Parenchymal necrosis - death and loss of hepatocytes
- Fibrous scarring (fibrosis) - diffuse, bridging fibrosis replacing normal architecture
- Regenerative nodules - islands of surviving hepatocytes attempting regeneration, surrounded and distorted by fibrosis
- Hepatocellular dysfunction (failure of liver's synthetic, metabolic, and detoxification functions)
- Portal hypertension (distorted intrahepatic vasculature increases resistance to portal flow)
2. ETIOLOGY
| Cause | Notes |
|---|---|
| Alcohol (Alcoholic Liver Disease) | Most common in Western countries; threshold ~80 g ethanol/day for 10+ years (women more susceptible at lower doses); Laennec's cirrhosis |
| Chronic Hepatitis C (HCV) | Major global cause; typically 20-30 years from infection to cirrhosis |
| Chronic Hepatitis B (HBV) | Most prevalent globally; especially in Asia and Africa; accelerated with HBV-HIV co-infection |
| Non-Alcoholic Fatty Liver Disease (NAFLD/NASH) | Rapidly becoming the most common cause in developed countries; linked to metabolic syndrome |
| Autoimmune Hepatitis (AIH) | Anti-smooth muscle antibody (ASMA, anti-actin), ANA positive; responds to immunosuppression |
| Primary Biliary Cholangitis (PBC) | Anti-mitochondrial antibody (AMA M2) positive; predominantly middle-aged women; bile duct destruction |
| Primary Sclerosing Cholangitis (PSC) | Stricturing fibro-inflammatory disease of bile ducts; strongly associated with ulcerative colitis; "beads on a string" on MRCP |
| Hereditary Hemochromatosis | Iron overload (HFE gene mutation - C282Y homozygous); "bronze diabetes"; affects liver, pancreas, heart, joints |
| Wilson's Disease | Copper accumulation (ATP7B mutation); Kayser-Fleischer rings; low serum ceruloplasmin; young patients |
| α1-Antitrypsin Deficiency | PAS-positive globules accumulate in hepatocytes; protease-antiprotease imbalance → cirrhosis + emphysema |
| Cardiac Cirrhosis | Prolonged right heart failure → chronic hepatic venous congestion → "nutmeg liver" → fibrosis |
| Budd-Chiari Syndrome | Hepatic vein thrombosis → centrizonal necrosis → fibrosis |
| Drugs/Toxins | Methotrexate, amiodarone, isoniazid, methyldopa, vitamin A (hypervitaminosis) |
| Cryptogenic | ~5-10%; often unrecognized burnt-out NASH |
3. PATHOGENESIS
Step-by-Step Mechanism
- Repeated cycles of hepatocyte injury and death from any of the above causes
- Necroinflammation: Kupffer cells (hepatic macrophages) and recruited lymphocytes release pro-inflammatory cytokines (TNF-α, IL-1β, IL-6)
- Normally, hepatic stellate cells reside in the space of Disse in a quiescent state, storing vitamin A (retinoids)
- Injured hepatocytes and activated Kupffer cells release TGF-β1 (transforming growth factor-β1) - the master profibrotic cytokine - plus PDGF, endothelin-1, and reactive oxygen species
- TGF-β1 transforms quiescent stellate cells into myofibroblasts (express α-smooth muscle actin - α-SMA)
- Activated myofibroblasts:
- Produce massive amounts of collagen types I and III → fibrosis
- Produce MMP inhibitors (TIMPs) → prevent collagen degradation
- Contract (via α-SMA) → sinusoidal narrowing → increased intrahepatic resistance
- Bridging fibrosis (porto-portal and porto-central bridges) divides liver into regenerative nodules
- Micronodular cirrhosis (nodules <3 mm): typically alcohol, hemochromatosis, biliary cirrhosis
- Macronodular cirrhosis (nodules >3 mm): typically post-viral (HCV, HBV), Wilson's disease
- Mixed: both types present
- Normal hepatic sinusoids have fenestrated endothelium (allows exchange between blood and hepatocytes) and no basement membrane
- In cirrhosis, sinusoids develop a continuous basement membrane and lose their fenestrations ("capillarization") → impaired hepatocyte-blood exchange → metabolic dysfunction even before hepatocyte death
- Fibrosis + regenerative nodules + stellate cell contraction → increased intrahepatic resistance → elevated portal venous pressure
- Normal portal pressure: 5-10 mmHg; HVPG (hepatic venous pressure gradient) ≤5 mmHg
- Clinically significant portal hypertension (complications begin): HVPG ≥10-12 mmHg
- Portal hypertension → splanchnic arteriolar vasodilation (nitric oxide, prostacyclin) → "underfilling" → RAAS and SNS activation → Na⁺/water retention → ascites and edema (see diagram)
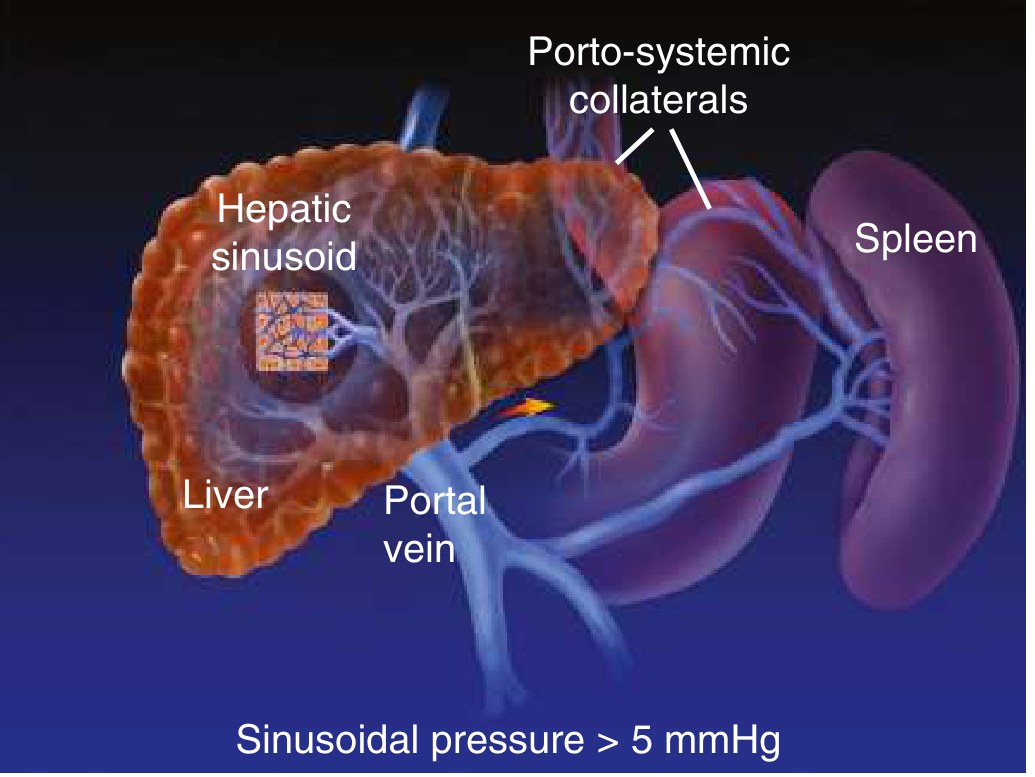
4. HISTOLOGICAL CLASSIFICATION
By Nodule Size
| Type | Nodule Size | Common Causes |
|---|---|---|
| Micronodular (Laennec's) | <3 mm; uniform small nodules | Alcohol, hemochromatosis, biliary cirrhosis, cardiac |
| Macronodular (post-necrotic) | >3 mm; irregular large nodules | Chronic viral hepatitis (HCV, HBV), Wilson's, AIH |
| Mixed | Both small and large nodules | Long-standing disease, any cause |
Histological Features
- Diffuse fibrosis: bands of collagen (types I and III) separating parenchyma
- Regenerative nodules: surrounded by fibrous septa
- Inflammatory infiltrate: lymphocytes, plasma cells
- Hepatocyte changes: ballooning, necrosis, steatosis, Mallory-Denk bodies (alcohol)
- Vascular changes: arterialized sinusoids, obliterated central veins, aberrant vessels
- Bile duct proliferation: "ductular reaction"
- Activation markers: α-SMA positive cells (activated stellate cells) in portal tracts and septa
Fibrosis Staging (Metavir / Ishak Score)
| Stage | Fibrosis |
|---|---|
| F0 | No fibrosis |
| F1 | Portal fibrosis without septa |
| F2 | Few septa |
| F3 | Many septa without cirrhosis (bridging fibrosis) |
| F4 | Cirrhosis |
5. CLINICAL FEATURES
A. Compensated Cirrhosis
- Often asymptomatic - discovered incidentally during imaging or blood tests
- Nonspecific symptoms: fatigue, weakness, weight loss, decreased muscle mass (sarcopenia), decreased libido, sleep disturbances
- ~40% already have esophageal varices (non-bleeding = compensated)
- Median survival: >12 years; annual mortality ~1-3.4%
B. Physical Signs of Chronic Liver Disease
- Jaundice, scleral icterus (from ↓ bilirubin excretion)
- Pallor (anemia - multifactorial)
- Scratch marks (pruritus from bile salt accumulation - especially cholestatic cirrhosis)
- Wasting, muscle loss (sarcopenia - very important prognostic marker)
- Leukonychia (white nails, Terry's nails) - hypoalbuminemia
- Clubbing - chronic hypoxia (hepatopulmonary syndrome)
- Dupuytren's contracture - palmar fibrosis; strongly associated with alcohol
- Palmar erythema - hyperestrogenism (↑ estrogen from impaired hepatic metabolism)
- Asterixis ("liver flap") - coarse tremor of hands with wrists extended; sign of hepatic encephalopathy; due to brief, sudden lapses in sustained posture (negative myoclonus)
- Parotid enlargement (alcohol)
- Xanthelasma (periocular yellow deposits in cholestatic cirrhosis/PBC)
- Fetor hepaticus: musty, sweet breath odor from mercaptans (dimethyl sulfide); indicates portosystemic shunting
- Spider angiomata (spider nevi): dilated central arteriole with radiating "legs"; >5 in the drainage area of the SVC = significant; from hyperestrogenism + ↑ substance P
- Gynecomastia (in males - hyperestrogenism)
- Loss of chest/axillary hair (hyperestrogenism)
- Testicular atrophy (hyperestrogenism + direct gonadotoxicity of alcohol)
- Caput medusae: dilated periumbilical veins (portosystemic collateral - portal → paraumbilical → epigastric superficial veins); flow is away from umbilicus
- Ascites: shifting dullness (>1.5 L), fluid thrill (>3 L), bulging flanks
- Splenomegaly: congestive splenomegaly from portal hypertension
- Liver: initially enlarged and firm (alcoholic, fatty), eventually shrunken, hard, irregular in advanced cirrhosis
- Peripheral edema (hypoalbuminemia + Na⁺ retention)
- Hepatic hydrothorax: right-sided (usually) pleural effusion from passage of ascitic fluid through diaphragmatic defects
C. Decompensated Cirrhosis
- Ascites (most common - 80% at time of decompensation)
- Variceal hemorrhage (esophageal or gastric)
- Hepatic encephalopathy
- Jaundice
- Spontaneous bacterial peritonitis (SBP)
- Hepatorenal syndrome (HRS)
6. COMPLICATIONS OF CIRRHOSIS (DETAILED)
A. PORTAL HYPERTENSION
| Collateral Site | Portal Tributary → Systemic Vein | Clinical Manifestation |
|---|---|---|
| Gastroesophageal junction | Left gastric → azygous (via submucosal esophageal veins) | Esophageal varices (most dangerous) |
| Fundus of stomach | Short gastric → left renal vein | Gastric varices (bleed more severely) |
| Umbilicus | Paraumbilical → epigastric veins | Caput medusae |
| Anus/rectum | Superior rectal → inferior rectal | Anorectal varices/hemorrhoids |
| Retroperitoneum | Mesenteric → retroperitoneal veins | Abdominal varices (incidental finding) |
B. ASCITES
- Portal hypertension → splanchnic arteriolar vasodilation (via NO, prostacyclin)
- ↓ Effective arterial blood volume ("underfilling") → baroreceptor activation
- Activation of RAAS, SNS, and ADH → renal Na⁺ and water retention
- Salt retention precedes ascites formation; RAAS activation is an early event even when GFR is normal
- As liver function worsens: ↓ albumin synthesis → ↓ oncotic pressure → ascitic fluid accumulates
- Hepatic lymph production exceeds thoracic duct capacity → lymph weeps from liver surface into peritoneum
- SAAG = Serum albumin - Ascites albumin
- SAAG ≥1.1 g/dL: Portal hypertension (cirrhosis, CHF, Budd-Chiari)
- SAAG <1.1 g/dL: Non-portal hypertension cause (malignancy, TB, pancreatitis, nephrotic syndrome)
- Grade 1: Only detectable by ultrasound
- Grade 2: Moderate - evident on clinical examination (bulging flanks, shifting dullness)
- Grade 3: Large - tense ascites; easily detectable; diaphragmatic splinting → dyspnoea
| Parameter | Cirrhosis (Transudative) | SBP | Malignant | TB Peritonitis |
|---|---|---|---|---|
| Appearance | Clear/straw | Turbid/cloudy | Bloody/turbid | Turbid/cloudy |
| Total protein | <2.5 g/dL | Variable | >2.5 g/dL | >2.5 g/dL |
| SAAG | ≥1.1 | ≥1.1 | <1.1 | <1.1 |
| WBC | <250 cells/µL | >250 PMN/µL | Variable | Lymphocyte predominant |
| Culture | Negative | +ve (E. coli, Klebsiella) | - | AFB culture (low yield) |
| Cytology | Negative | - | Positive (50-70%) | Negative |
C. SPONTANEOUS BACTERIAL PERITONITIS (SBP)
- Bacterial translocation from gut lumen → mesenteric lymph nodes → systemic circulation → ascitic fluid
- Most common organisms: Gram-negative bacteria (E. coli most common, then Klebsiella pneumoniae) → 70%; Gram-positive (Streptococcus pneumoniae, Enterococcus) → 30%
- Risk factors: low ascitic protein (<1.5 g/dL) + low complement in ascitic fluid → reduced opsonic activity
- Intestinal bacterial overgrowth (decreased GI motility in cirrhosis) promotes translocation
- Abdominal pain/tenderness (60%)
- Fever (80%) - may be low-grade
- Deteriorating hepatic encephalopathy
- Silent SBP (30-40%) - no symptoms; detected only by routine diagnostic paracentesis
- Acute kidney injury (complication)
- Diagnostic paracentesis: PMN >250 cells/mm³ = treat empirically
- Blood cultures (50% positive); ascitic fluid culture in blood culture bottles
- IV Cefotaxime 2 g every 8 hours × 5 days (drug of choice)
- IV Albumin (1.5 g/kg on day 1 + 1 g/kg on day 3) - prevents hepatorenal syndrome (reduces 3-month mortality from 33% to 10% in SORT trial)
- Ciprofloxacin/Ofloxacin IV: alternative if resistant organisms unlikely
- Primary prophylaxis: Norfloxacin 400 mg/day in high-risk patients (ascitic protein <1.5 g/dL + Child C, or renal impairment, or hyponatremia)
- Secondary prophylaxis (after first SBP episode): Norfloxacin 400 mg/day indefinitely OR ciprofloxacin 500 mg/day; reduces SBP recurrence from 68% to 20% at 1 year
D. HEPATIC ENCEPHALOPATHY (HE)
- Ammonia is the central toxin: normally detoxified in liver via urea cycle → in cirrhosis, reduced hepatic mass + portosystemic shunting → ammonia accumulates in systemic circulation → crosses blood-brain barrier
- Ammonia → astrocyte swelling (Alzheimer type II astrocytosis) → cerebral edema
- Ammonia → upregulates peripheral benzodiazepine receptors (on astrocytes) → ↑ neurosteroid synthesis (allopregnanolone) → GABA-A receptor potentiation → cortical depression
- Manganese accumulates in globus pallidus → motor dysfunction
- Inflammatory state (cytokines, sepsis) dramatically worsens HE
| Grade | Features |
|---|---|
| 0 (MHE - Minimal) | Subclinical; only detected by neuropsychological tests (impaired driving, impaired memory) |
| 1 | Mild confusion, altered sleep rhythm, euphoria or mild anxiety; asterixis subtle |
| 2 | Drowsiness; lethargic; moderate confusion; obvious asterixis; inappropriate behavior |
| 3 | Somnolent but arousable; marked confusion; disoriented to time and place; asterixis |
| 4 | Coma (unresponsive to verbal or painful stimuli) |
- Fluid/electrolyte disturbance: hypokalemia (↑ renal NH₃ production), metabolic alkalosis, hyponatremia
- Alcohol excess / Azotemia (renal failure → ↑ urea → ↑ ammonia)
- Constipation / Carbohydrate excess
- Toxins (sedatives, opioids, benzodiazepines - avoid these in cirrhosis)
- Sepsis (SBP, pneumonia, UTI) / Surgery / GI bleed (large protein load in gut → ↑ ammonia)
- Identify and treat precipitants (most important step)
- Lactulose (first-line): 30-60 mL 2-3 times daily; titrate to 2-3 soft stools/day
- Mechanism: acidifies colonic pH → traps NH₄⁺ (ionized, non-absorbable); also cathartic effect; reduces transit time; alters gut flora
- Rifaximin (550 mg twice daily): non-absorbable antibiotic; reduces gut bacterial metabolism of nitrogenous compounds; reduces recurrence by 58% (RIFT trial)
- Branched-chain amino acids (BCAA): IV infusion benefits patients with HE; competes with aromatic amino acids for brain uptake
- Correct hypokalemia, metabolic alkalosis
- Avoid benzodiazepines, opioids, sedatives
- Protein intake: do NOT restrict protein (worsens sarcopenia) - target 1.2-1.5 g/kg/day
- TIPS patients: increased HE risk; may need prophylactic rifaximin
E. VARICEAL HEMORRHAGE
- Gastroesophageal varices present in 50% at cirrhosis diagnosis; prevalence: Child A 40% → Child C 85%
- Growth rate: 7-8% per year
- Annual bleeding risk: small varices ~5%, large varices ~15%
- Mortality per bleed: 10-15% (Rosen's Emergency Medicine)
- High-risk stigmata: large varices + red wale markings + Child C
F. HEPATORENAL SYNDROME (HRS)
- Extreme splanchnic vasodilation → profound renal vasoconstriction → dramatically reduced renal blood flow → oliguria + rising creatinine
- The kidneys are structurally normal (if transplanted to a healthy recipient, they function normally)
- Any precipitant (SBP, large-volume paracentesis without albumin, nephrotoxic drugs, GI bleed) can trigger HRS in decompensated cirrhosis
| Type | Course | Prognosis |
|---|---|---|
| HRS-AKI (formerly Type 1) | Rapid (doubling of creatinine to >2.5 mg/dL in <2 weeks); acute and rapidly progressive | Very poor - median survival <2 weeks without treatment |
| HRS-CKD (formerly Type 2) | Slowly progressive; moderate renal failure; often in setting of refractory ascites | Months; bridge to transplant |
- Cirrhosis + ascites
- Serum creatinine rising ≥0.3 mg/dL in 48 hours OR ≥50% from baseline within 7 days
- No improvement after 2 days of albumin challenge (1 g/kg IV albumin) + diuretic withdrawal
- Absence of shock; no nephrotoxic drugs
- No parenchymal kidney disease (no proteinuria >500 mg/day, no hematuria, no abnormal renal ultrasound)
- Terlipressin + IV albumin (1 g/kg/day, max 100 g/day): vasoconstricts splanchnic bed → improves renal perfusion; reverses HRS in 40-50%
- Norepinephrine + IV albumin: equivalent to terlipressin; used in ICU
- TIPS: can improve HRS type 2 (refractory ascites); bridge to transplant
- Liver transplantation: only definitive cure; kidney function normalizes after transplant in most cases
- Avoid nephrotoxic drugs, NSAIDs, aminoglycosides, contrast media
G. HEPATOPULMONARY SYNDROME AND PORTOPULMONARY HYPERTENSION
- Pulmonary capillary dilation (up to 500 µm vs normal 8 µm) → intrapulmonary shunt → hypoxemia
- Classic sign: platypnoea (dyspnoea that worsens when upright, improves when supine) + orthodeoxia (SaO₂ falls on standing)
- Diagnosis: contrast echocardiography (bubble echo) - delayed appearance of bubbles in left heart (>3 cycles)
- Treatment: liver transplantation (reverses HPS in most cases)
- Pulmonary arterial hypertension in setting of portal hypertension
- Vasoconstrictive substances reaching pulmonary bed via portosystemic shunts
- Diagnosis: mean pulmonary artery pressure >25 mmHg + elevated PVR on right heart catheterization
- Treatment: pulmonary vasodilators (sildenafil, bosentan); liver transplant in mild-moderate POPH (contraindicated if mPAP >50 mmHg)
H. HEPATOCELLULAR CARCINOMA (HCC)
- Cirrhosis is the most important risk factor for HCC (all causes)
- Annual incidence of HCC in cirrhosis: 1-5%/year (highest in HBV, HCV, hemochromatosis)
- Surveillance: 6-monthly liver ultrasound ± AFP in all cirrhotic patients (reduces HCC-related mortality by 37%)
- AFP >200 ng/mL (in cirrhotic patient with arterial enhancement on imaging) = HCC until proven otherwise
- Treatment depends on Barcelona Clinic Liver Cancer (BCLC) staging: surgical resection, ablation, TACE, TIPS, sorafenib, lenvatinib, liver transplantation (Milan criteria)
I. COAGULOPATHY IN CIRRHOSIS
- Liver produces all coagulation factors EXCEPT vWF and Factor VIII (both produced by endothelium)
- Cirrhosis → ↓ all hepatically-synthesized coagulation factors (I, II, V, VII, IX, X, XI) → ↑ PT/INR
- Also: ↓ protein C, S, antithrombin → pro-thrombotic tendency despite elevated INR (a "rebalanced hemostasis")
- Elevated INR/thrombocytopenia is NOT a contraindication to paracentesis
- Before invasive procedures: target platelet >50,000/mm³; use cryoprecipitate (not FFP) for active bleeding correction
J. HEPATIC HYDROTHORAX
- Right-sided pleural effusion (usually) in cirrhosis without cardiopulmonary disease
- Pathogenesis: ascitic fluid passes through small diaphragmatic defects into pleural space
- Diagnosis: SAAG of pleural fluid mirrors ascites (both have SAAG ≥1.1)
- Management: diuretics (spironolactone + furosemide); avoid thoracentesis unless dyspnoeic; TIPS in refractory cases
7. INVESTIGATIONS
Laboratory
| Test | Finding in Cirrhosis | Significance |
|---|---|---|
| ALT, AST | Mildly elevated or normal (advanced cirrhosis - burnt-out); AST:ALT >2 → alcohol | Necroinflammation marker |
| ALP, GGT | Elevated (especially in cholestatic disease); GGT markedly ↑ in alcohol | Cholestasis |
| Bilirubin | Elevated (marker of decompensation) | Hepatic synthetic/excretory failure |
| Albumin | Reduced (<3.5 g/dL) | Chronic synthetic failure (half-life 20 days) |
| PT/INR | Prolonged | Acute synthetic failure (Factor VII shortest half-life) |
| FBC | Thrombocytopenia (splenomegaly), anemia (multifactorial), leukopenia | Hypersplenism |
| Serum Na⁺ | Hyponatremia (<130 mEq/L in 25%) | Poor prognosis marker |
| Creatinine/BUN | Elevated → HRS, volume depletion | Renal function |
| Serum ammonia | Elevated in HE (but does not correlate well with grade) | Encephalopathy marker |
| AFP | Elevated → HCC surveillance | >200 ng/mL = diagnostic threshold |
Specific Etiological Tests
| Etiology | Tests |
|---|---|
| Alcohol | GGT:ALP ratio; MCV (macrocytosis); CDT (carbohydrate-deficient transferrin); history |
| HBV | HBsAg, anti-HBc, HBe Ag/Ab, HBV DNA |
| HCV | Anti-HCV antibody, HCV RNA, genotype |
| Autoimmune hepatitis | ANA, ASMA (anti-F-actin), anti-LKM1; raised IgG |
| PBC | Anti-mitochondrial antibody (AMA-M2), raised IgM, ALP >5× ULN |
| PSC | MRCP ("beads on a string"), ANCA; associated IBD |
| Hemochromatosis | Serum ferritin, transferrin saturation ≥45%, HFE gene mutation (C282Y) |
| Wilson's disease | Serum ceruloplasmin (<20 mg/dL), 24-hr urine copper (>100 µg/day), Kayser-Fleischer rings (slit-lamp) |
| α1-AT deficiency | Serum α1-antitrypsin level, phenotype (PIZZ most severe) |
Imaging
- Liver ultrasound (first-line): nodular liver surface, increased echogenicity, splenomegaly, ascites, portal vein diameter (>13 mm suggests portal hypertension), hepatic veins patent/occluded
- Doppler ultrasound: portal vein flow direction (hepatofugal = severe portal hypertension), portal vein velocity
- CT abdomen (triphasic): assess for HCC (arterial enhancement + portal washout), splenomegaly, varices, ascites
- MRI liver: superior soft tissue contrast; hepatic iron quantification (hemochromatosis)
- MRCP: biliary tree evaluation (PSC)
- FibroScan (transient elastography): liver stiffness in kPa; non-invasive fibrosis quantification; cutoff ~12-14 kPa for cirrhosis
- Upper GI endoscopy (OGD): esophageal/gastric varices, portal hypertensive gastropathy
Liver Biopsy
- Gold standard for confirming cirrhosis, staging fibrosis, and identifying etiology
- Percutaneous (Menghini/Trucut needle) or transjugular (when coagulopathy is significant)
- Sampling error is a limitation (liver biopsy assesses only 1/50,000 of total liver volume)
- Non-invasive alternatives (FibroScan, serum biomarkers) increasingly preferred for staging
8. SEVERITY SCORING
Child-Turcotte-Pugh (CTP) Score
| Parameter | 1 Point | 2 Points | 3 Points |
|---|---|---|---|
| Ascites | None | Grade 1-2/easily treated | Grade 3-4/refractory |
| Hepatic encephalopathy | None | Grade 1-2/precipitant-induced | Grade 3-4/spontaneous |
| Bilirubin (mg/dL) | <2 | 2-3 | >3 |
| Albumin (g/dL) | >3.5 | 2.8-3.5 | <2.8 |
| PT prolongation (sec) / INR | <4 / <1.7 | 4-6 / 1.7-2.3 | >6 / >2.3 |
| Class | Score | 1-Year Survival | 2-Year Survival |
|---|---|---|---|
| A | 5-6 | 100% | 85% |
| B | 7-9 | 81% | 57% |
| C | 10-15 | 45% | 35% |
MELD Score (Model for End-Stage Liver Disease)
- Range 6-40; higher = worse prognosis
- MELD ≥15: benefit from liver transplantation outweighs risk
- MELD-Na (modified): incorporates serum sodium (adds further prognostic power)
- Used for organ allocation priority on transplant waiting lists
9. MANAGEMENT
A. General Measures (All Patients)
-
Treat the underlying cause - most important intervention:
- Alcohol: complete abstinence (most effective in alcoholic cirrhosis; fibrosis can regress)
- HBV: Tenofovir (TDF/TAF) or Entecavir indefinitely → regression of cirrhosis in 75% at 5 years (Sleisenger & Fordtran)
- HCV: Direct-acting antivirals (DAAs) - cure rates >95%; fibrosis regression possible
- NASH: Weight loss (even 5-10% reduces fibrosis), SGLT2 inhibitors, GLP-1 agonists, resmetirom
- Hemochromatosis: Phlebotomy (remove 500 mL blood/week); deferoxamine for those who can't tolerate venesection
- Wilson's disease: D-penicillamine or trientine + zinc
-
Nutrition:
- Protein: 1.2-1.5 g/kg/day (DO NOT restrict protein - worsens sarcopenia and HE)
- Calorie intake: 35-40 kcal/kg/day
- Late-evening snack (branched-chain amino acids) - reduces nocturnal catabolism in cirrhosis
- Sodium restriction: 2000 mg (88 mmol)/day for ascites management
- Avoid excess vitamin A, herbal medicines, hepatotoxic drugs (NSAIDs, alcohol)
-
Vaccinations: Hepatitis A, Hepatitis B (if not immune), pneumococcal, influenza, COVID-19
-
Avoid:
- NSAIDs (worsen renal function, precipitate HRS)
- ACE inhibitors / ARBs in decompensated cirrhosis (↓BP + worsen renal function - Rosen's EM)
- Sedatives, opioids, benzodiazepines (precipitate HE)
- Aminoglycosides (nephrotoxic)
- Contrast media (use with caution; pre-hydrate)
B. Management of Ascites
- Diuretics (first-line pharmacological therapy):
- Spironolactone 100 mg + Furosemide 40 mg (5:2.5 ratio) once daily
- Titrate upward every 3-5 days maintaining the ratio: max spironolactone 400 mg + furosemide 160 mg
- Rationale: spironolactone blocks aldosterone (the major driver of Na⁺ retention in cirrhosis); furosemide prevents hyperkalemia from spironolactone
- Fluid restriction only if Na⁺ <125 mEq/L
- Target weight loss: 0.5 kg/day (ascites alone) or 1 kg/day (ascites + edema) - to prevent intravascular depletion
- Check urine Na/K ratio: >1 suggests non-adherence to sodium restriction; <1 suggests diuretic resistance
- Large-volume paracentesis (LVP): 4-6 L at one sitting
- Must give IV albumin 8 g per litre of ascitic fluid removed when >5 L removed - prevents post-paracentesis circulatory dysfunction (PPCD) and HRS
- Note: INR elevation and thrombocytopenia are NOT contraindications to paracentesis (Rosen's EM)
- TIPS (Transjugular Intrahepatic Portosystemic Shunt):
- Metallic stent connecting portal vein to hepatic vein within liver parenchyma
- Effectively lowers portal pressure → reduces ascites production
- Contraindications: severe HE, advanced Child C (CTP >13), active infection, severe pulmonary hypertension, severe right heart failure
- Complication: hepatic encephalopathy (20-30%) - portosystemic shunting bypasses liver
- Liver transplantation (definitive)
C. Management of SBP
D. Management of Hepatic Encephalopathy
E. Management of Variceal Hemorrhage
- Non-selective beta-blockers (NSBB): Propranolol 40 mg BD (titrate to max tolerated; target HR 55-60 bpm) OR Carvedilol 6.25 mg BD (preferred; also reduces intrahepatic resistance via α-blockade)
- Endoscopic variceal band ligation (EVL): if NSBB intolerant; repeat sessions every 2-4 weeks until variceal obliteration
- NSBB + EVL combination NOT superior to either alone for primary prophylaxis
- Resuscitate: transfuse to Hb 7-8 g/dL (restrictive strategy - lower portal pressure)
- Vasoactive drugs (start before endoscopy, continue for 3-5 days):
- Terlipressin (first-line): 1-2 mg IV every 4-6 hours - vasopressin analogue → splanchnic vasoconstriction
- Alternative: Octreotide (somatostatin analogue) 50 µg bolus then 50 µg/hour infusion
- Prophylactic IV antibiotics (mandatory): IV Ceftriaxone 1 g/day × 5-7 days (or norfloxacin if low-risk) - reduces SBP, re-bleeding, and mortality
- Urgent endoscopy (EVL) within 12 hours - band ligation of esophageal varices
- Sengstaken-Blakemore / Minnesota tube (balloon tamponade): temporary hemostasis only, maximum 24 hours, as bridge to definitive treatment
- TIPS: for refractory or early re-bleed; "Early TIPS" (within 72 hours) in high-risk patients (Child B with active bleeding, Child C <14)
- NSBB + repeated EVL sessions (combination superior to either alone)
- NSBB: propranolol or carvedilol indefinitely
F. Management of Hepatorenal Syndrome
G. Liver Transplantation
- MELD ≥15 with liver-related complications
- Child-Pugh C
- Refractory ascites
- Recurrent HE
- Refractory variceal bleeding
- HRS (especially Type 1)
- HCC within Milan criteria (single lesion ≤5 cm, or ≤3 lesions each ≤3 cm, no vascular invasion, no extrahepatic spread)
- SBP (marker of very poor prognosis without transplant)
- Active alcohol/drug use (require ≥6 months sobriety)
- Extrahepatic malignancy
- Severe cardiopulmonary disease (portopulmonary hypertension with mPAP >50 mmHg)
- Active uncontrolled sepsis
- Anatomical barriers to transplantation
- Deceased donor liver transplantation (DDLT) - orthotopic (OLT)
- Living donor liver transplantation (LDLT) - right lobe from living donor
- Split liver transplantation
10. PROGNOSIS AND NATURAL HISTORY
| Stage | Annual Mortality | Events |
|---|---|---|
| Compensated cirrhosis | 1-3.4%/year | Varices develop 7-8%/year; progression to decompensation 5-7%/year |
| Decompensation (first event) | 15-20%/year | Second decompensation greatly worsens prognosis |
| Acute-on-chronic liver failure (ACLF) | 30-90% (30-day) | Acute deterioration + organ failure(s); most severe |
- MAP <82 mmHg (independent predictor of mortality - Rosen's EM)
- MELD ≥18-20
- Hyponatremia (<130 mEq/L)
- HCC development
- Bacterial infections (dramatically worsen short-term mortality)
- Sarcopenia and frailty
- Previously considered irreversible, but anti-fibrotic effects of treating the underlying cause are well documented:
- HBV treated with TDF: cirrhosis regression in 75% at 5 years
- HCV cured with DAAs: significant fibrosis regression in many patients
- Alcohol abstinence: meaningful improvement in early cirrhosis; advanced Child C may not reverse
- Bariatric surgery / weight loss in NASH: histological improvement
- Goldman-Cecil Medicine (Portal hypertension pathophysiology, ascites mechanism, encephalopathy, decompensated cirrhosis, Child-Pugh/MELD scores)
- Sleisenger & Fordtran's Gastrointestinal and Liver Disease (HBV treatment, nucleoside analogues, cirrhosis regression)
- Rosen's Emergency Medicine (Clinical pearls: SBP, HRS, paracentesis safety, terlipressin, TIPS)
- Henry's Clinical Diagnosis and Management (Etiology, non-invasive fibrosis markers, histology)
- Robbins & Kumar Basic Pathology (Stellate cell activation, fibrosis mechanism, nodule types)