## PATIENT SUMMARY FOR CLINICAL REVIEW## Baseline Medical History * Cardiovascular: Chronic Heart Disease. History of CABG (2006) and 1 Coronary Stent (2019). Severe Acute Decompensated Heart Failure episode ~1.5 years ago where Ejection Fraction (EF) was 20%. Most recent Echocardiogram (6 months ago) showed long-term improvement of EF to 30%. Managed with baseline Lasix, water restriction, and other cardiac medications. * Metabolic: Diabetes Mellitus (DM), Hypertension (HTN), and Hypercholesterolemia. * Renal: Chronic Kidney Disease (CKD) with a baseline Creatinine of 1.5–1.7. History of multiple prior AKI episodes peaking around 2.5. * Gastrointestinal / Infectious: Persistent perianal fistula previously treated with multiple seton valves. Currently awaiting an outpatient pelvic MRI once stabilized. ## Timeline of Current Admission * 2 Days Before Admission: Routine outpatient labs showed mild transaminase elevations with AST and ALT both in the early 100s. * Day of Admission: Admitted with suspected Myocardial Infarction (MI). Initial Troponin I was mildly elevated at 141. Main presentation was severe, unyielding nausea and vomiting. LFTs were not repeated upon admission. Continuous IV Heparin drip was initiated. * Day 2 of Admission (Yesterday): Serial Troponin I results remained flat (neither rising nor falling); acute MI was officially ruled out. IV Heparin drip was permanently stopped. * Day 3 of Admission (Today - Active Crisis): * GI Symptoms: Severe, unyielding nausea and vomiting have persisted without improvement. * Liver Function Tests (LFTs): Massive, acute spike noted. ALT is 2500 and AST is 4500. (Ammonia level was checked and returned normal). * Coagulation Profile (Off Heparin): * D-Dimer: Critically elevated at 10,377 ng/mL * INR: Elevated at 2.79 * Prothrombin Time (PT): Prolonged at 32.4 seconds * aPTT: Near-normal at 38.7 seconds * Vital Signs & Neuro Status Shifts: * Blood pressure and oxygen saturation (98–99%) have remained consistently normal. * Body temperature suddenly dropped to a hypothermic 93°F (33.9°C); patient reports feeling cold. * Mental status has shifted to a waxing and waning pattern; he can engage in normal conversation but frequently drifts into periods of drowsiness. * Breathing pattern changed: Baseline breathing is normal, but periodically interrupted by sudden, slightly labored breaths that cause visible abdominal shaking. ( explain the diagnosis and reason for it. )
Clinical Analysis: Day 3 Crisis
1. The Massive LFT Spike (ALT 2500 / AST 4500): Ischemic Hepatitis ("Shock Liver")
Diagnosis
Why This Patient?
"Of all cases of extreme serum AST elevations (to >3000 U/L), ischemic hepatitis accounts for about half. The most common cause of ischemic hepatitis is cardiovascular disease, which accounts for more than 70% of cases... More than 80% of cases of ischemic hepatitis occur in the setting of heart failure."
"The presence of heart failure significantly increases the likelihood that a drop in cardiac output from any cause will result in ischemic hepatitis."
Why is the term "ischemic hepatitis" actually a misnomer?
"Because hepatitis refers to inflammation of the liver, the term ischemic hepatitis is a misnomer, because inflammation is typically not present. A more physiologic term would be hypoxic hepatitis, because the primary cause of this syndrome is tissue hypoxia, which may be the result of hypoperfusion from cardiac failure or shock... The characteristic pathologic feature is acute centrilobular necrosis."
Why Did the LFTs Spike AFTER Admission?
"Serum aminotransferase levels peak 1 to 3 days after the hemodynamic insult and return to normal within 7 to 10 days."
The AST > ALT Pattern (AST 4500 vs ALT 2500)
2. The Coagulopathy: Deranged INR/PT with Near-Normal aPTT + Massively Elevated D-Dimer
What is Happening
- INR 2.79 / PT 32.4 sec - prolonged
- aPTT 38.7 sec - near-normal
- D-Dimer 10,377 ng/mL - critically elevated
"Chronic liver disease is characterized by the impaired synthesis of coagulation factors, resulting in prolongation of the prothrombin time (PT) and INR."
The Massively Elevated D-Dimer (10,377)
- Hepatic failure impairs clearance of fibrin degradation products - the liver normally clears activated clotting factors and FDPs from circulation
- Low-grade DIC can accompany severe acute liver injury and systemic hypoperfusion
- The heparin was stopped - the heparin was actually partially masking coagulopathic activity; now off heparin, the coagulation picture is unmasked
- His history of heart failure with low EF promotes venous stasis and micro-thrombus formation with subsequent fibrinolysis - all of which generate D-dimer
3. The Hypothermia (93°F / 33.9°C)
4. Waxing and Waning Mental Status
Diagnosis: Hepatic Encephalopathy (HE) - ammonia-independent mechanism
- Reduced hepatic clearance of endogenous toxins beyond ammonia - including aromatic amino acids, mercaptans, false neurotransmitters, and GABA-like compounds that accumulate when the liver fails
- Systemic hypoperfusion and reduced cerebral perfusion - with an EF of 30% at best (likely acutely worse), cardiac output may be insufficient for adequate cerebral perfusion, particularly during any positional or hemodynamic fluctuation
- Metabolic derangement from the combination of acute liver failure, renal insufficiency (his baseline CKD with AKI risk), and the systemic stress response
- Cheyne-Stokes induced hypoxia (see below) - periodic hypoventilation causes recurrent CO2 retention and hypoxia that directly impairs consciousness during the apneic phases
5. The Periodic Breathing Pattern (Sudden Labored Breaths with Abdominal Shaking): Cheyne-Stokes Respiration
Diagnosis: Cheyne-Stokes Respiration
The Mechanism (from Guyton and Hall Textbook of Medical Physiology):
"When a long delay occurs for transport of blood from the lungs to the brain, changes in CO2 and O2 in the alveoli can continue for many more seconds than usual. Under these conditions, the storage capacities of the alveoli and pulmonary blood for these gases are exceeded; then, after a few more seconds, the periodic respiratory drive becomes extreme and Cheyne-Stokes breathing begins. This type of Cheyne-Stokes breathing often occurs in patients with severe cardiac failure because blood flow is slow, thus delaying the transport of blood gases from the lungs to the brain."
- Low cardiac output causes a prolonged lung-to-brain circulation time (normally ~5-10 seconds, extended to 30-60+ seconds in severe HF)
- When the CO2 level rises in the blood, ventilation increases - but by the time the newly oxygenated, CO2-depleted blood reaches the brain's respiratory center, too much time has passed
- The respiratory center now sees a sudden CO2 drop and over-suppresses breathing - causing apnea or hypopnea
- CO2 then rises again in the blood, the respiratory center eventually sees it, and fires a burst of deep, labored breathing - this is the visible abdominal heaving the team is observing
- The cycle repeats
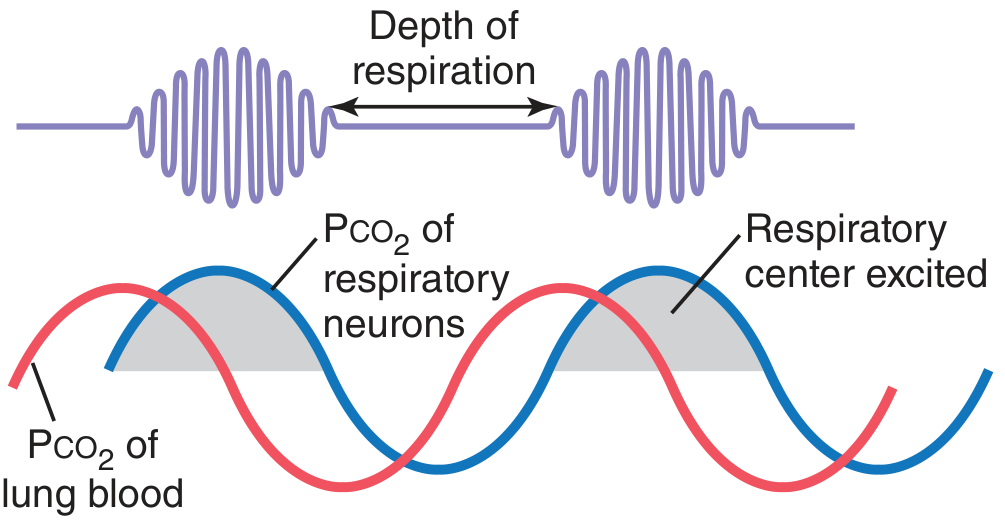
"Cheyne-Stokes breathing is seen in chronic heart failure, as well as in stroke and traumatic brain injury." - Fishman's Pulmonary Diseases and Disorders
Unified Pathophysiologic Picture
SEVERE DECOMPENSATED HEART FAILURE (EF acutely worsened from 30%)
|
┌───────────────┼───────────────┐
↓ ↓ ↓
↓Hepatic perfusion ↓Cardiac output ↓Cerebral perfusion
| | |
Centrilobular Prolonged Encephalopathy
hepatic necrosis lung-to-brain (waxing/waning)
| circulation time |
ALT 2500 / | |
AST 4500 Cheyne-Stokes ↓
| respiration Hypothermia
| (periodic labored (liver = major
↓Coag factor breathing) heat source,
synthesis now failing)
INR 2.79/
PT 32.4
|
Impaired FDP
clearance →
D-Dimer 10,377
Sources
- Sleisenger and Fordtran's Gastrointestinal and Liver Disease - "Ischemic Hepatitis," p. 2580-2613
- Tietz Textbook of Laboratory Medicine, 7th Edition - "Ischemic Hepatitis (Shock Liver)," block 22
- Guyton and Hall Textbook of Medical Physiology - "Cheyne-Stokes Breathing," p. 539 (Fig. 42.12)
- Fishman's Pulmonary Diseases and Disorders - "Disturbances of Respiratory Rhythm"
- Morgan and Mikhail's Clinical Anesthesiology, 7e - "Coagulation in Liver Disease"