Atherosclerosis
Reading File
Finding Sources
Searching Images
atherosclerosis plaque formation pathology
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Now I have comprehensive information from Robbins Pathology and Guyton & Hall. Let me compile a thorough response.
Atherosclerosis
Definition
Atherosclerosis is a disease of large and intermediate-sized arteries characterized by intimal lesions called atheromas (atherosclerotic plaques) — raised lesions with soft, friable lipid cores (mainly cholesterol and cholesterol esters with necrotic debris) covered by fibrous caps. It is best understood as the vascular response to endothelial injury.
It underlies the pathogenesis of coronary, cerebral, and peripheral vascular disease, and accounts for roughly half of all deaths in the Western world.
— Robbins & Kumar Basic Pathology, p. 314
Pathogenesis
Step 1 — Endothelial Injury & Dysfunction
The initiating event is damage to the vascular endothelium (from hyperlipidemia, hypertension, cigarette toxins, diabetes, hemodynamic shear stress, etc.). Injured endothelium:
- Upregulates adhesion molecules
- Loses ability to produce nitric oxide (a vasodilator and anti-adhesion factor)
- Becomes permeable to circulating lipoproteins
Step 2 — Lipid Accumulation & Foam Cell Formation
Circulating monocytes and LDL particles accumulate at injury sites. Monocytes cross the endothelium, differentiate into macrophages, and ingest oxidized LDL via scavenger receptors — producing foam cells. Aggregates of foam cells form visible fatty streaks, the earliest atherosclerotic lesion.
Step 3 — Chronic Inflammation
Cholesterol crystals and free fatty acids trigger the inflammasome, producing IL-1 and other cytokines. Activated T cells secrete IFN-γ, further activating macrophages, endothelial cells, and smooth muscle cells (SMCs).
Step 4 — SMC Proliferation & Plaque Growth
Growth factors (PDGF, FGF, TGF-α) from platelets, macrophages, and endothelial cells drive SMC migration from the media into the intima, where they proliferate and synthesize extracellular matrix (ECM). This forms the fibrous cap. Fatty streaks coalesce and grow into full atheromatous plaques.
Step 5 — Plaque Complications
- Stenosis: Large plaques mechanically reduce luminal flow
- Calcification: Calcium precipitates into plaques → arterial rigidity ("hardening of the arteries")
- Rupture → Thrombosis: Plaque rupture exposes the thrombogenic lipid core to blood → acute thrombus → sudden vessel occlusion (MI, stroke)
- Aneurysm formation: Ischemia and inflammation weaken the underlying media
— Guyton and Hall Textbook of Medical Physiology, p. 851; Robbins & Kumar Basic Pathology, pp. 314–318
Plaque Progression (Guyton & Hall diagram)
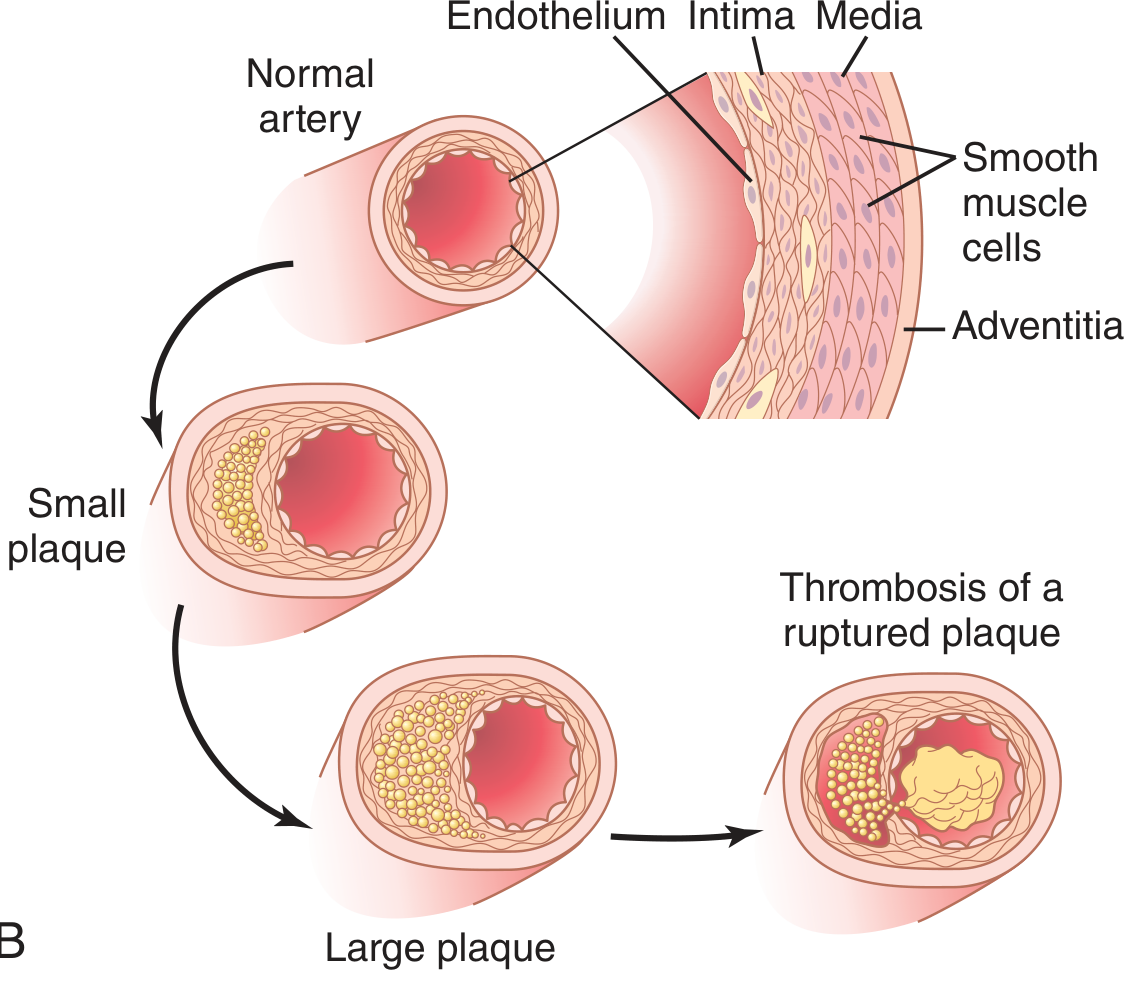
Risk Factors
| Non-Modifiable (Constitutional) | Modifiable |
|---|---|
| Family history / genetic predisposition | Hyperlipidemia (esp. elevated LDL) |
| Increasing age | Hypertension |
| Male sex | Cigarette smoking |
| Mendelian disorders (e.g., familial hypercholesterolemia) | Diabetes mellitus |
| Inflammation (elevated CRP) |
Risk factors are multiplicative: hyperlipidemia + hypertension + smoking together increase MI risk ~7-fold.
Premenopausal women have relative protection (though clinical trials have NOT confirmed a benefit from post-menopausal estrogen replacement).
— Robbins & Kumar Basic Pathology, p. 315–317
Vulnerable (Unstable) Plaques
Not all plaques are equally dangerous. Vulnerable plaques — those most prone to rupture — are characterized by:
- Large lipid cores
- Thin fibrous caps with few SMCs
- Dense inflammatory infiltrates (foam cells, T lymphocytes)
- Active collagen degradation (via matrix metalloproteinases from macrophages)
Critically, plaques responsible for acute MI are often not severely stenotic before the event — they rupture without warning. Extrinsic triggers include adrenergic surges (e.g., waking, emotional stress), which explain the circadian peak of MI onset between 6 AM and noon.
Statins stabilize plaques both by lowering LDL and by reducing plaque inflammation, independent of lipid-lowering effects.
— Robbins & Kumar Basic Pathology, p. 318
Preferred Sites
Atherosclerosis preferentially affects areas of turbulent flow and low shear stress:
- Abdominal aorta (more than thoracic)
- Coronary arteries
- Popliteal arteries
- Carotid bifurcation
- Circle of Willis
Dyslipidemia Mechanism
- Elevated LDL → oxidized LDL in the intima → macrophage scavenger receptor uptake → foam cells
- Oxidized LDL is cytotoxic to ECs and SMCs, and promotes cytokine/chemokine release
- High HDL is protective: reverse cholesterol transport, anti-inflammatory effects
- Lowering LDL (diet or statins) slows progression and can regress plaques
Clinical Consequences
| Site | Consequence |
|---|---|
| Coronary arteries | Angina, myocardial infarction |
| Cerebral arteries / carotid | TIA, ischemic stroke |
| Peripheral arteries | Claudication, limb ischemia |
| Renal arteries | Renovascular hypertension |
| Mesenteric arteries | Bowel ischemia |
| Aorta | Aortic aneurysm, dissection |
This is a shared conversation. Sign in to Orris to start your own chat.