Acquired pancytopenia
cd /home/daytona/workspace/attachments/34497369-f4b9-4f8c-affe-9c55cd3baa1c && python3 -c " import pdfplumber import re pdf_path = 'Part XIX -Diseases of the Blood - Nelson 2024.pdf' with pdfplumber.open(pdf_path) as pdf: print(f'Total pages: {len(pdf.pages)}') # Search for pancytopenia for i, page in enumerate(pdf.pages): text = page.extract_text() or '' if 'pancytopenia' in text.lower() or 'aplastic' in text.lower(): print(f'--- Page {i+1} ---') print(text[:3000]) print() " 2>&1 | head -300
cd /home/daytona/workspace/attachments/34497369-f4b9-4f8c-affe-9c55cd3baa1c && python3 << 'EOF' import pdfplumber pdf_path = 'Part XIX -Diseases of the Blood - Nelson 2024.pdf' with pdfplumber.open(pdf_path) as pdf: print(f'Total pages: {len(pdf.pages)}') for i in range(min(5, len(pdf.pages))): text = pdf.pages[i].extract_text() or '' print(f'--- Page {i+1} (chars: {len(text)}) ---') print(text[:500]) print() EOF
cd /home/daytona/workspace/attachments/34497369-f4b9-4f8c-affe-9c55cd3baa1c && python3 << 'EOF' import pdfplumber pdf_path = 'Part XIX -Diseases of the Blood - Nelson 2024.pdf' with pdfplumber.open(pdf_path) as pdf: print(f'Total pages: {len(pdf.pages)}') # Search for pancytopenia in all pages hits = [] for i, page in enumerate(pdf.pages): text = page.extract_text() or '' if 'pancytopenia' in text.lower(): hits.append((i+1, text)) print(f'Pages with pancytopenia: {[h[0] for h in hits]}') # Print each hit for pg, text in hits[:6]: print(f'\n=== PAGE {pg} ===') print(text[:3000]) EOF
cd /home/daytona/workspace/attachments/34497369-f4b9-4f8c-affe-9c55cd3baa1c && python3 << 'EOF' import pdfplumber pdf_path = 'Part XIX -Diseases of the Blood - Nelson 2024.pdf' with pdfplumber.open(pdf_path) as pdf: # Search pages 1-80 for pancytopenia/aplastic anemia for i in range(80): text = pdf.pages[i].extract_text() or '' if 'pancytopenia' in text.lower() or 'aplastic anemia' in text.lower(): print(f'\n=== PAGE {i+1} ===') print(text[:2500]) EOF
cd /home/daytona/workspace/attachments/34497369-f4b9-4f8c-affe-9c55cd3baa1c && python3 << 'EOF' import pdfplumber pdf_path = 'Part XIX -Diseases of the Blood - Nelson 2024.pdf' with pdfplumber.open(pdf_path) as pdf: total = len(pdf.pages) print(f"Total: {total}") # Try pages 50-80 only for i in range(50, 80): text = pdf.pages[i].extract_text() or '' if 'pancytopenia' in text.lower() or 'aplastic' in text.lower(): print(f'\n=== PAGE {i+1} ===') print(text[:2000]) EOF
cd /home/daytona/workspace/attachments/34497369-f4b9-4f8c-affe-9c55cd3baa1c && python3 << 'EOF' import pdfplumber pdf_path = 'Part XIX -Diseases of the Blood - Nelson 2024.pdf' with pdfplumber.open(pdf_path) as pdf: # Focus on pages 80-161 for i in range(80, 161): text = pdf.pages[i].extract_text() or '' if 'pancytopenia' in text.lower() or 'aplastic' in text.lower(): print(f'\n=== PAGE {i+1} ===') print(text[:2500]) EOF
cd /home/daytona/workspace/attachments/34497369-f4b9-4f8c-affe-9c55cd3baa1c && python3 << 'EOF' import pdfplumber pdf_path = 'Part XIX -Diseases of the Blood - Nelson 2024.pdf' with pdfplumber.open(pdf_path) as pdf: # Pages 100-130 for pancytopenia for i in range(100, 130): text = pdf.pages[i].extract_text() or '' if 'pancytopenia' in text.lower() or ('aplastic' in text.lower() and 'anemia' in text.lower()): print(f'\n=== PAGE {i+1} ===') print(text[:2500]) EOF
cd /home/daytona/workspace/attachments/34497369-f4b9-4f8c-affe-9c55cd3baa1c && python3 << 'EOF' import pdfplumber pdf_path = 'Part XIX -Diseases of the Blood - Nelson 2024.pdf' with pdfplumber.open(pdf_path) as pdf: # Pages 25-50 for i in range(20, 55): text = pdf.pages[i].extract_text() or '' if 'pancytopenia' in text.lower() or ('aplastic' in text.lower()): print(f'\n=== PAGE {i+1} ===') print(text[:2500]) EOF
cd /home/daytona/workspace/attachments/34497369-f4b9-4f8c-affe-9c55cd3baa1c && python3 << 'EOF' import pdfplumber pdf_path = 'Part XIX -Diseases of the Blood - Nelson 2024.pdf' with pdfplumber.open(pdf_path) as pdf: # Pages 130-161 - end of book for aplastic anemia for i in range(125, 161): text = pdf.pages[i].extract_text() or '' if 'pancytopenia' in text.lower() or 'aplastic' in text.lower() or 'acquired' in text.lower(): print(f'\n=== PAGE {i+1} ===') print(text[:3000]) EOF
cd /home/daytona/workspace/attachments/34497369-f4b9-4f8c-affe-9c55cd3baa1c && python3 << 'EOF' import pdfplumber pdf_path = 'Part XIX -Diseases of the Blood - Nelson 2024.pdf' with pdfplumber.open(pdf_path) as pdf: # Check pages 7-22 for aplastic anemia chapter for i in range(6, 23): text = pdf.pages[i].extract_text() or '' if 'aplastic' in text.lower() or 'acquired' in text.lower(): print(f'\n=== PAGE {i+1} ===') print(text[:3000]) EOF
Acquired Pancytopenia
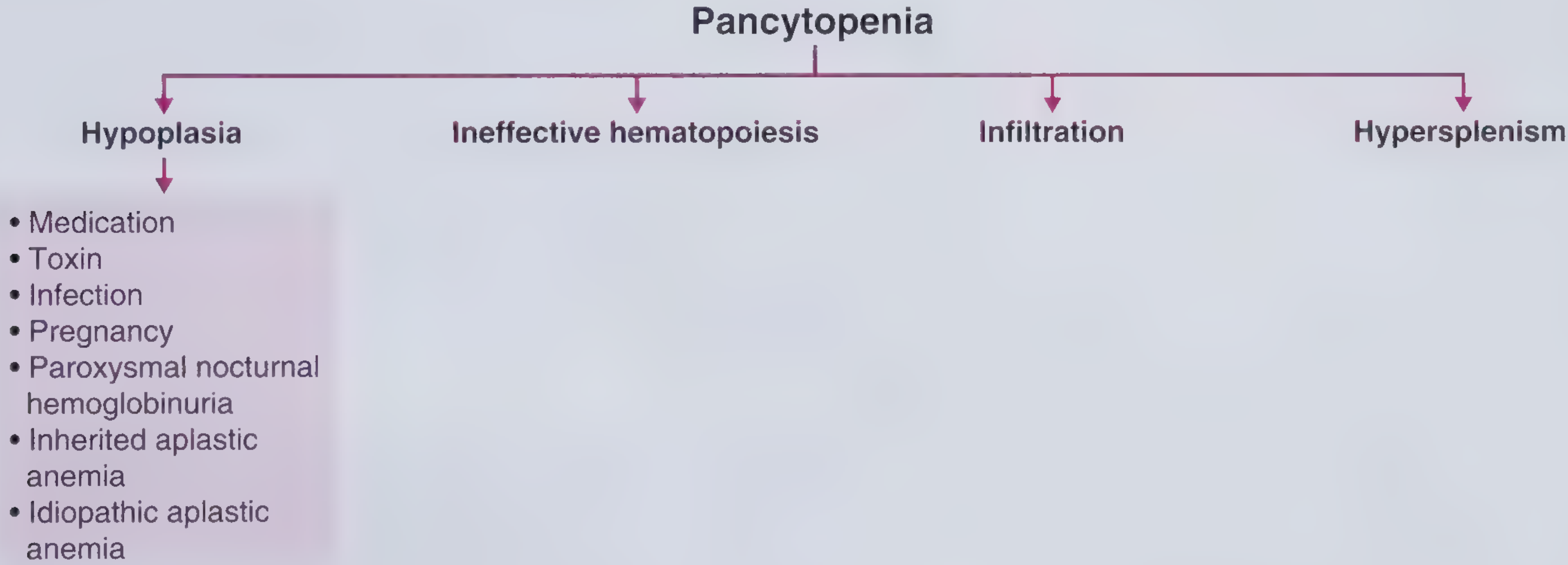
Classification by Bone Marrow Findings (Harrison's 22e)
1. Pancytopenia with Hypocellular (Aplastic) Marrow
- Acquired (immune) aplastic anemia
- Hypoplastic myelodysplastic syndrome (MDS)
- Aleukemic leukemia (rare)
- Some acute lymphoid leukemia
- Copper deficiency
2. Pancytopenia with Cellular Marrow
| Primary Bone Marrow Diseases | Secondary to Systemic Disease |
|---|---|
| Myelodysplastic syndromes (MDS) | Systemic lupus erythematosus (SLE) |
| Paroxysmal nocturnal hemoglobinuria (PNH) | Hypersplenism |
| Myelofibrosis | Vitamin B12/folate deficiency |
| Aleukemic leukemia | Copper deficiency |
| Myelophthisis | Alcohol |
| Bone marrow lymphoma | HIV infection |
| Hairy cell leukemia | Brucellosis, TB, Leishmaniasis |
| Sarcoidosis, Sepsis |
3. Hypocellular Marrow without Full Pancytopenia
- Q fever, Legionnaires' disease, Anorexia nervosa/starvation, Mycobacterium
Acquired Aplastic Anemia (Primary Focus)
Epidemiology
- Incidence: 2 per million/year in Europe and Israel; 5-7 per million/year in Thailand and China
- Bimodal age distribution: major peak in teens/twenties, second peak in older adults
- Equal sex distribution
Etiology
- Definite (dose-dependent): Cytotoxic chemotherapy agents (antimetabolites, antimitotics, some antibiotics), benzene
- Idiosyncratic reactions (unpredictable): Chloramphenicol, NSAIDs (phenylbutazone, indomethacin, ibuprofen), anticonvulsants (hydantoins, carbamazepine), heavy metals (gold, arsenic), sulfonamides, antithyroid drugs (methimazole, PTU), antihistamines (cimetidine), d-penicillamine, antidiabetics (tolbutamide), allopurinol, methyldopa, quinidine, carbamazepine, lithium, phenothiazines
- Seronegative hepatitis (non-A, B, C): Accounts for ~5% of AA cases; typically young men, severe aplasia 1-2 months after hepatitis; likely immune-mediated
- EBV (infectious mononucleosis): Rarely causes AA
- Parvovirus B19: Causes pure red cell aplasia (transient aplastic crisis in chronic hemolytic anemias); rarely generalized marrow failure
- HIV-1: Pancytopenia via marrow infiltration and immunosuppression
- Eosinophilic fasciitis (rare collagen vascular syndrome)
- Thymoma and hypoimmunoglobulinemia
- SLE
- Transfusion-associated GVHD (nonirradiated blood products to immunodeficient recipient)
- Large granular lymphocytosis (LGL syndrome)
- CTLA4 deficiency
- PNH clones are detectable by flow cytometry in ≥50% of AA patients at presentation
- Up to 50% of PNH patients develop AA; conversely, ~50% of AA patients have small PNH clones
- Classic triad: hemolysis (Coombs-negative), thrombosis, and pancytopenia/marrow failure
- Thrombosis is a major cause of morbidity (Budd-Chiari, portal/splenic vein, cerebral)
Pathophysiology of Acquired AA (Immune-Mediated)
- Activated cytotoxic T-cell clones (oligoclonal, expanded) are found in blood and marrow - they decline with successful immunosuppression
- Type 1 cytokines are produced: interferon-gamma (IFN-γ) induces Fas (CD95) expression on CD34+ stem cells, triggering apoptosis
- CD34+ cells are reduced to ≤1% of normal in severe disease
- HLA loss on HSCs allows immune escape and PNH clone expansion
- Genetically determined features (HLA polymorphisms, cytokine gene variants, T-cell regulatory gene variants) determine why only some individuals exposed to a trigger develop AA
Other Acquired Causes
Myelodysplastic Syndromes (MDS)
Bone Marrow Infiltration (Myelophthisis)
- Malignancy: acute leukemia, lymphoma, multiple myeloma, metastatic carcinoma (breast, prostate, lung, stomach)
- Infection: miliary tuberculosis (caseating granulomas on biopsy; pancytopenia mostly in HIV+ patients), fungi, brucellosis
- Fibrosis: primary myelofibrosis, or secondary (myelophthisis) from above conditions
- Storage diseases: Gaucher disease
- Classic finding: "dry tap" on aspiration; leukoerythroblastic blood picture (tear-drop cells, nucleated RBCs, immature myeloid cells)
Hypersplenism
- Splenomegaly causing sequestration and premature destruction of blood cells
- Massive spleens can sequester up to 90% of platelets, 65% of granulocytes, and 30% of RBCs
- Bone marrow is normo- or hypercellular (reactive)
- Causes: cirrhosis/portal hypertension, myeloproliferative disease, lymphoma, infections, storage diseases
- Splenectomy can be curative when hypersplenism is the sole driver
Vitamin B12/Folate Deficiency
Drug-Induced Marrow Suppression
Other Toxins
- Alcohol: direct marrow toxicity + folate deficiency; may persist despite cessation
- Arsenic poisoning
- Benzene (industrial solvent exposure)
Severity Classification of Aplastic Anemia
| Criteria | Severe AA | Very Severe AA |
|---|---|---|
| Marrow cellularity | < 25% or <50% with <30% residual cells | same |
| Neutrophils | < 0.5 × 10⁹/L | < 0.2 × 10⁹/L |
| Platelets | < 20 × 10⁹/L | same |
| Reticulocytes | < 20 × 10⁹/L (absolute) | same |
Clinical Features
- Bleeding: Most common early symptom - easy bruising, gum oozing, epistaxis, heavy menses, petechiae; risk of intracranial hemorrhage with severe thrombocytopenia
- Anemia symptoms: Lassitude, weakness, dyspnea, palpitations
- Infection: Not the usual first symptom (unlike agranulocytosis), but neutropenic fever becomes a major complication
- Absent organomegaly: Patients often look surprisingly well despite very low counts; absence of lymphadenopathy and hepatosplenomegaly helps distinguish from malignancy
- Seronegative hepatitis AA: abrupt presentation in young male following recent hepatitis
Diagnosis
- CBC: Pancytopenia; macrocytosis common; absolute reticulocyte count low
- Peripheral smear: No dysplastic cells (unlike MDS), no blasts (unlike leukemia), no schistocytes
- Bone marrow biopsy (required): Hypocellular marrow with fat replacement; residual lymphocytes and plasma cells; no fibrosis or infiltration
- Bone marrow aspirate: May be "dry tap" in severe cases
- Chromosomal analysis: Normal karyotype in immune AA (abnormalities suggest MDS/leukemia); chromosomal breakage studies if Fanconi anemia is suspected
- Flow cytometry: PNH clone detection (GPI-anchor-deficient RBCs and granulocytes) - should be performed in all patients
- LFTs/hepatitis serology: Relevant if seronegative hepatitis suspected
- Vitamin B12, folate, copper levels
- Autoimmune workup (ANA, dsDNA) if SLE suspected
Treatment
Definitive Therapy
- Treatment of choice for young patients (<40 years, or up to 50 if suitable) with severe/very severe AA and a matched sibling donor
- Cures marrow failure; eliminates risk of clonal evolution
- Preferred over immunosuppression if profound neutropenia in younger patients
- Patients who fail immunosuppression can be salvaged with SCT later
- Overall response rate 70-80%; complete response ~50%
- Horse ATG is significantly superior to rabbit ATG
- Mechanism of ATG: depletes autoreactive T-cells; early serendipitous observation of immune pathophysiology
- Cyclosporine: oral, titrated by blood levels; side effects include nephrotoxicity, hypertension, seizures
- Eltrombopag (TPO mimetic): once daily for 6 months; likely stimulates HSC directly; side effect is hepatotoxicity
- Serum sickness (~day 10 of ATG): flu-like illness with skin eruption and arthralgias - treated with methylprednisolone
- Relapse is common (as cyclosporine/eltrombopag tapered); most patients respond to reinstitution
- Clonal evolution to MDS or leukemia occurs in ~10-15% over a decade
- Unverified in controlled trials but some patients respond
- Upregulate telomerase activity (useful in telomere biology disorders)
- 3-4 month trial appropriate for moderate disease, especially if telomere defect present
Supportive Care
- Infections: Prompt empirical broad-spectrum IV antibiotics for neutropenic fever; anti-fungal therapy for persistent fever (Candida, Aspergillus); granulocyte transfusions for refractory bacterial/fungal infections
- Platelet transfusions: Maintain >10,000/μL prophylactically; use single-donor platelets; HLA-matched for alloimmunized patients; suppress menstruation with estrogens
- Red cell transfusions: Maintain Hb ≥70 g/L (~2 units every 2 weeks); iron chelation (deferoxamine or deferasirox) after ~50th transfusion
- Growth factors (G-CSF, EPO): Generally NOT effective in immune aplastic anemia
- Avoid aspirin and NSAIDs (thrombocytopenia)
Pediatric Perspective (Nelson 2024)
- Acquired aplastic anemia (immune-mediated)
- Acute leukemia (blasts on smear/marrow - key diagnostic distinction)
- Myelodysplastic syndrome
- PNH (Chapter 510 Nelson 2024 - rare in children; classic presentation includes pancytopenia/aplastic anemia preceding or following PNH diagnosis)
- Hemophagocytic lymphohistiocytosis (HLH)
- Vitamin B12/folate deficiency
- Infections (EBV, CMV, parvovirus B19, HIV, TB, visceral leishmaniasis)
- SLE
Key References
- Harrison's Principles of Internal Medicine 22E (2025), Chapter 107, pp. 850-856
- Nelson Textbook of Pediatrics 2024 (Part XIX - Diseases of the Blood), Chapters 496, 510, 517
- Frameworks for Internal Medicine, Chapter 27 (Pancytopenia), pp. 358-365
- Robbins, Cotran & Kumar: Pathologic Basis of Disease (aplastic anemia section)
Only from Nelson
cd /home/daytona/workspace/attachments/34497369-f4b9-4f8c-affe-9c55cd3baa1c && python3 << 'EOF' import pdfplumber pdf_path = 'Part XIX -Diseases of the Blood - Nelson 2024.pdf' with pdfplumber.open(pdf_path) as pdf: # Chapter 517 and 518 on pancytopenias - find those pages for i in range(55, 100): text = pdf.pages[i].extract_text() or '' if ('517' in text or '518' in text) and ('aplastic' in text.lower() or 'pancytopenia' in text.lower()): print(f'\n=== PAGE {i+1} ===') print(text[:3000]) EOF
Acquired pancytopenia - from Nelson only
cd /home/daytona/workspace/attachments/34497369-f4b9-4f8c-affe-9c55cd3baa1c && python3 << 'EOF' import pdfplumber pdf_path = 'Part XIX -Diseases of the Blood - Nelson 2024 (1).pdf' with pdfplumber.open(pdf_path) as pdf: print(f'Total pages: {len(pdf.pages)}') # Find Chapter 518 - Acquired Pancytopenias for i in range(90, 110): text = pdf.pages[i].extract_text() or '' if 'acquired pancytopenia' in text.lower() or '518' in text: print(f'\n=== PAGE {i+1} ===') print(text[:4000]) EOF
cd /home/daytona/workspace/attachments/34497369-f4b9-4f8c-affe-9c55cd3baa1c && python3 << 'EOF' import pdfplumber pdf_path = 'Part XIX -Diseases of the Blood - Nelson 2024 (1).pdf' with pdfplumber.open(pdf_path) as pdf: # Get ALL of pages 94-97 (Chapter 518) in full for i in range(93, 98): text = pdf.pages[i].extract_text() or '' print(f'\n========== PAGE {i+1} ==========') print(text) EOF
Acquired Pancytopenias
Definition
- Pancytopenia with hypocellular marrow - inherited bone marrow failure syndromes (IBMFSs), acquired aplastic anemia, or hypoplastic MDS
- Pancytopenia with cellular marrow - infiltrative or replacement processes, MDS with ineffective hematopoiesis
- Hypocellular marrow without full pancytopenia
Etiology and Epidemiology
- Direct destruction of hematopoietic progenitors
- Disruption of the marrow microenvironment
- Immune-mediated suppression of marrow elements
Table 518.1 - Etiology of Acquired Aplastic Anemia
- Predictable: Chemotherapy, benzene
- Idiosyncratic: Chloramphenicol, antiepileptics, gold, MDMA ("ecstasy"), NSAIDs, antibiotics
- Cytomegalovirus (CMV)
- Epstein-Barr virus (EBV)
- Hepatitis B, Hepatitis C
- Hepatitis non-A, non-B, non-C (seronegative hepatitis)
- HIV
- COVID-19
- Eosinophilic fasciitis
- Hypoimmunoglobulinemia
- Thymoma
- Common variable immunodeficiency syndrome (NFKB1)
- Pregnancy
- Paroxysmal nocturnal hemoglobinuria (PNH)
- Leukemia
- Myelodysplasia
- Myelofibrosis
- Autoimmune diseases
- Vitamin B12 deficiency
- Folate deficiency
- Copper deficiency
- Cryptic dyskeratosis congenita (no physical stigmata)
- Telomerase reverse transcriptase haploinsufficiency
- Atypical presentation of genetic marrow failure syndromes
- Leishmaniasis
Drug and Toxin Details (Table 518.2)
- Antineoplastic agents: fluorouracil, mercaptopurine, 6-thioguanine, methotrexate, cytosine arabinoside, gemcitabine, fludarabine, cladribine, pentostatin, hydroxyurea; busulfan, cyclophosphamide, chlorambucil, nitrogen mustard, melphalan, cisplatin, carboplatin, ifosfamide, nitrosoureas, mitomycin C; daunorubicin, doxorubicin, mitoxantrone; vinblastine, paclitaxel; etoposide
- Antimicrobials: chloramphenicol, dapsone, fluorocytosine
- Antiinflammatory: colchicine
- Insecticides: chlordane, DDT, lindane, parathion
- Other chemicals: benzene, kerosene, chlorophenols, carbon tetrachloride
- Antimicrobials: chloramphenicol, dapsone, sulfonamides, tetracycline, methicillin, amphotericin, quinacrine, chloroquine, pyrimethamine
- Anticonvulsants: hydantoins, carbamazepine, phenacemide, primidone, ethosuximide
- Antiinflammatory: phenylbutazone, indomethacin, ibuprofen, oxyphenbutazone, sulindac, naproxen
- Antiarrhythmic: quinidine, tocainide, procainamide
- Metals: gold, arsenic, mercury, bismuth
- Antihistamines: cimetidine, ranitidine, chlorpheniramine, pyrilamine, tripelennamine
- Diuretics: acetazolamide, furosemide, chlorothiazide, methazolamide
- Hypoglycemic agents: chlorpropamide, tolbutamide
- Antithyroid drugs: propylthiouracil, potassium perchlorate, methylthiouracil, methimazole, carbimazole
- Antihypertensive agents: methyldopa, enalapril, captopril
- Sedatives: chlordiazepoxide, chlorpromazine, meprobamate, prochlorperazine
Notable Specific Causes
- Drugs: Most notable are benzene, chloramphenicol, gold, and MDMA ("ecstasy")
- Parvovirus B19: Classically causes isolated RBC aplasia; can produce transient pancytopenia in patients with sickle cell disease or immunodeficiency
- Hepatitis viruses, herpesviruses, EBV, CMV, HIV: Can cause prolonged pancytopenia
- Immune-mediated AA: Seronegative hepatitis, eosinophilic fasciitis, and thymoma are specifically implicated
- PNH and collagen vascular diseases: Must be evaluated
- Bone marrow replacement: Leukemia, neuroblastoma (classically), myelofibrosis, MDS, osteopetrosis - importantly, hypoplastic anemia can precede acute leukemia by a few months
- Many acquired cases are idiopathic - no causative agent identified; most are probably immune-mediated via cytotoxic T lymphocytes and cytokine destruction
Pathology and Pathogenesis
Severity Classification
- ≥2 cell components seriously compromised:
- Absolute neutrophil count (ANC) < 500/mm³
- Platelet count < 20,000/mm³
- Reticulocyte count < 1% (after correction for hematocrit)
- Bone marrow biopsy with < 30% cellularity
- ANC 500-1,500/mm³
- Platelet count 20,000-100,000/mm³
- Reticulocyte count < 1%
- Importantly, ~65% of patients presenting with moderate disease will eventually progress to severe disease if simply observed
Mechanisms of Bone Marrow Failure
- Direct cytotoxic effect on HSCs from a drug or chemical
- Cell-mediated or antibody-dependent cytotoxicity
- Immune-mediated: Most idiopathic cases - increased circulating cytotoxic T lymphocytes producing interferon-γ (IFN-γ) that suppress hematopoiesis
- Early apoptosis: Abnormal telomere length and telomerase activity in granulocytic precursors suggest early apoptosis of hematopoietic progenitors may play a role
Associated Cytogenetic Abnormalities
- Uniparental disomy of 6p
- Monosomy 7 / deletion of 7q
- Trisomy 8, 6, or 15
Associated Genes
Clinical Manifestations, Laboratory Findings, and Differential Diagnosis
- Cardiac failure
- Fatigue
- Infection
- Bleeding
Differential Diagnosis to Consider
- Cancers (leukemia, neuroblastoma)
- Collagen vascular disorders
- PNH (may respond to specific therapy)
- Infections - especially parvovirus (responds to IV immunoglobulin)
Diagnostic Workup
- Peripheral blood smear - examine RBC, leukocyte, and platelet morphology
- Reticulocyte count - to assess erythropoietic activity
- Chromosomal breakage analysis - mandatory in children to evaluate for Fanconi anemia (Chapter 517)
- Telomere length - to evaluate for telomeropathies
- Fetal hemoglobin level - elevated suggests congenital pancytopenia (not diagnostic)
- Flow cytometry for CD55 and CD59 on erythrocytes and granulocytes - most sensitive test for PNH
- Bone marrow examination - both aspirate AND biopsy, evaluated for:
- Morphologic features
- Cellularity
- Cytogenetic abnormalities
Treatment
A. Hematopoietic Stem Cell Transplantation (HSCT)
- Offers 90% chance of long-term survival
- Preparative regimens typically consist of cyclophosphamide + fludarabine + horse ATG
- Alemtuzumab (anti-CD52 monoclonal antibody)-based conditioning also being evaluated
- Risks: graft failure, graft-versus-host disease, late effects (secondary cancers, cataracts, short stature, hypothyroidism, gonadal dysfunction)
- Only ~20% of patients have an HLA-matched family member donor
B. Immunosuppression (for patients without sibling donor)
- Response rate: 60-70%
- Median time to response: 6 months
- 30-60% of responders relapse after discontinuation of immunosuppression; some must continue cyclosporine for several years
- Among those who relapse: ~50% respond to a second course of ATG + cyclosporine
- Granulocyte CSF or GM-CSF sometimes added to ATG + cyclosporine for very severe neutropenia (ANC < 200/mm³)
- No clear evidence this influences response rate or survival
- Higher baseline reticulocyte count → higher probability of response and survival
- Shorter telomere length → higher probability of relapse
C. For Refractory or Relapsed Disease
- Matched-unrelated HSCT or T-cell-depleted haploidentical family member HSCT - response rate approaching 90%
- Cord blood transplants - survival ~90% in this refractory pediatric group
- Eltrombopag (oral TPO mimetic agent): Ongoing studies, shows promise in patients ≥15 years with refractory disease; produces hematologic response (improved platelets, neutrophils, and hemoglobin) in over half the patients, with trilineage normalization on marrow biopsy in some
- High-dose cyclophosphamide - used in patients not suitable for HSCT and inadequate response to immunosuppression; causes prolonged severe pancytopenia → high risk of life-threatening fungal infections
- Other therapies with inconsistent results: Androgens, corticosteroids, plasmapheresis
- Alemtuzumab as monotherapy in relapsed disease - improved response rates and 3-year survival vs additional ATG + cyclosporine
Complications
- Thrombocytopenic bleeding - life-threatening
- Neutropenic infections - serious bacterial infections AND invasive mycoses (especially after multiple courses of antibiotics)
- Alloantibodies to RBC antigens - from repeated transfusions
- Transfusional iron overload - requires iron chelation therapy in chronically transfused patients
Prognosis
- Spontaneous recovery from pancytopenia rarely occurs
- If left untreated, severe pancytopenia has:
- ~50% mortality within 6 months of diagnosis
- >75% overall mortality
- Infection and hemorrhage are the major causes of morbidity and mortality
- The majority of children with acquired severe aplastic anemia respond to allogeneic marrow transplantation or immunosuppression, achieving normal or near-normal blood cell counts
Pancytopenia Caused by Marrow Replacement
- Malignant infiltration: Neuroblastoma (classical), leukemia - important: hypoplastic anemia can precede acute leukemia by a few months, so serial monitoring is essential in apparent AA
- Myelofibrosis, MDS, osteopetrosis
- Morphologic examination of peripheral blood and bone marrow + cytogenetic studies are critical for distinguishing these
Myelodysplastic Syndromes (MDS) in Children
- Refractory cytopenia of childhood: Peripheral blasts < 2% and marrow blasts < 5%
- MDS with excess blasts: Peripheral blasts 2-19% and/or marrow blasts 5-19%
- Disease with > 20% blasts = AML
- One group: Somatic variants in > 25 genes leading to MDS
- Another group (younger patients, < 55 years): Autoimmune suppression by clonal expansion of T lymphocytes (similar to idiopathic AA)
- Must rule out medications, B12, folate, copper deficiencies as causes
- Allogeneic HSCT survival: ~60%
- Exception: MDS/AML in Down syndrome - very responsive to conventional chemotherapy, long-term survival > 80%
- Lenalidomide → best for 5q− abnormality
- ATG + cyclosporine → most effective for trisomy 8, especially if PNH clone present
- Imatinib → targets tyrosine kinase mutations in t(5;12) and del(4q12)
- Azacitidine and decitabine → DNA hypomethylating agents for cases without known molecular target