biochemical reason forthe clinical sign and symptoms of classical galactosemia
Reading File
Finding Sources
Searching PubMed
classical galactosemia biochemical pathophysiology
Reading File
Reading File
Reading File
Searching PubMed
galactosemia GALT pathophysiology
Reading Article
PMID: 38986816
Reading Article
PMID: 37702899
Finding Sources
Reading File
Reading File
I now have very rich, multi-source content. Let me compile the comprehensive answer.
Classical Galactosemia: Biochemical Basis of Clinical Signs and Symptoms
The Enzyme Defect and Metabolic Block
Classical galactosemia is an autosomal recessive inborn error of metabolism caused by loss-of-function mutations in the GALT gene (chromosome 9), encoding galactose-1-phosphate uridylyltransferase (GALT). The most common mutation is the Q188R substitution.
Normally, lactose from milk is cleaved into glucose + galactose by intestinal lactase. Galactose then enters the Leloir pathway:
Galactose
↓ [Galactokinase (GALK)]
Galactose-1-phosphate (Gal-1-P)
↓ [GALT] ← BLOCKED IN CLASSICAL GALACTOSEMIA
UDP-galactose + Glucose-1-phosphate
↓ [UDP-galactose 4'-epimerase (GALE)]
UDP-glucose
With GALT absent or severely deficient, Gal-1-P accumulates in virtually every tissue (liver, brain, kidney, lens, RBCs, gonads). Two toxic alternative metabolites also build up:
- Galactitol: galactose is reduced by aldose reductase → galactitol (a polyol that cannot be further metabolised and becomes trapped in tissues)
- Galactonate: an oxidized by-product of excess galactose
Additionally, there is a deficit of UDP-galactose, an essential precursor for glycoprotein and glycolipid synthesis (cell surface receptors, myelin).
Biochemical Mechanisms Behind Each Clinical Feature
1. Neonatal Presentation: Vomiting, Diarrhea, Failure to Thrive
Symptoms begin within days of first milk ingestion. The accumulation of Gal-1-P in hepatocytes and enterocytes disrupts normal carbohydrate metabolism and cellular function. Gal-1-P competitively inhibits phosphoglucomutase, impairing glycogenolysis and normal glucose release. Feeding intolerance (vomiting, diarrhea, bloating) results from the rapidly accumulating toxic metabolites.
2. Jaundice and Hepatomegaly (first week of life)
- Early hepatomegaly is caused primarily by fatty change (steatosis) - Gal-1-P disrupts normal lipid metabolism in hepatocytes.
- Prolonged or severe disease leads to widespread hepatic scarring resembling alcoholic cirrhosis - Gal-1-P accumulation causes direct hepatocyte toxicity, oxidative stress (greater oxidative stress is associated with low GALT activity), and possibly mitochondrial dysfunction (see below).
- Jaundice may appear to continue from physiologic neonatal jaundice, but is actually unconjugated + conjugated hyperbilirubinemia from hepatocellular damage.
- Coagulopathy (easy bruisability, bleeding) follows from impaired hepatic synthesis of clotting factors.
- Hypoalbuminemia, ascites, and anasarca develop as liver synthetic function deteriorates.
2024 Biochemical Update (PMID 38986816): Galactose-1-phosphate has been shown to directly inhibit cytochrome c oxidase (Complex IV) of the mitochondrial electron transport chain, causing mitochondrial dysfunction, reduced ATP synthesis, and reduced respiratory rates. This is a newly identified mechanism explaining the severity of hepatocellular injury beyond simple metabolite accumulation.
3. Cataracts
Galactose that cannot enter the Leloir pathway is reduced by aldose reductase in the lens to galactitol. Because galactitol cannot be further metabolised or transported out of the lens, it accumulates and is trapped. This creates a powerful osmotic gradient, drawing water into the lens cells. The lens absorbs water, swells, and opacification (cataract) develops - often first seen as characteristic "oil-drop" changes on slit-lamp examination. This is a direct, specific, and fully biochemically explained consequence of the metabolic block.
4. Intellectual Disability / Brain Damage
Several mechanisms converge on the brain:
| Mechanism | Biochemical basis |
|---|---|
| Gal-1-P accumulation | Direct neurotoxicity; elevated Gal-1-P in neuronal tissues disrupts membrane function and signalling |
| Deficiency of UDP-galactose | Impairs glycoprotein and glycolipid synthesis - including myelin and cell-surface receptors essential for CNS function |
| Galactitol in neurons | Elevated galactitol causes osmotic damage and has been correlated with neurological outcomes |
| Oxidative stress | Low GALT activity is associated with increased oxidative stress and membrane damage due to insufficient antioxidant protection |
| Mitochondrial dysfunction | Gal-1-P inhibits cytochrome c oxidase, depleting ATP in energy-demanding neurons |
Neuropathological findings include: loss of nerve cells, gliosis, and edema, especially in the dentate nuclei of the cerebellum and olivary nuclei of the medulla, with variable changes in the cerebral cortex and white matter. These explain the cognitive impairment, ataxia, and speech defects seen even in treated patients.
5. Aminoaciduria / Renal Fanconi Syndrome
Accumulation of Gal-1-P in renal tubular cells impairs the amino acid transporter on the proximal tubule. This produces a generalised proximal tubular dysfunction (renal Fanconi syndrome) with:
- Aminoaciduria
- Albuminuria and aciduria
- Glycosuria (glucose reabsorption failure)
- Metabolic acidosis
6. E. coli Septicemia (Neonatal)
There is an increased susceptibility to fulminant Gram-negative (E. coli) septicemia in the first days of life. The biochemical basis is impaired neutrophil bactericidal activity - Gal-1-P accumulation in leukocytes disrupts their killing function (oxidative burst and phagocytic clearance are compromised). This is a potentially life-threatening complication that occurs before the diagnosis is even suspected.
7. Premature Ovarian Failure (Long-term)
Even with early dietary treatment, most women with classical galactosemia develop premature ovarian insufficiency (POI). The mechanisms are:
- Defective glycosylation - UDP-galactose deficiency impairs glycoprotein synthesis, including glycosylation of FSH and its receptor, disrupting gonadotropin signalling
- Defective germ cell migration - galactose metabolites may impair embryonic germ cell migration to the developing gonads during fetal development, reducing the initial oocyte pool
- Direct gonadal toxicity of Gal-1-P in ovarian tissue (streak ovaries are found on histology)
- Notably, testicular function is not significantly affected in males
8. Ataxia and Speech Defects (Long-term, even on treatment)
These late neurological complications persist because:
- Cerebellar damage (dentate nuclei) occurs early and may not fully reverse
- The UDP-galactose deficit may have caused irreversible impairment of myelin and glycoprotein formation during critical developmental windows
- Endogenous galactose production (from turnover of glycoproteins/glycolipids) continues even on a galactose-free diet, meaning low-level Gal-1-P accumulation is never fully eliminated
Summary Diagram
GALT deficiency
│
├─→ Gal-1-P accumulates ────┬─→ Liver: steatosis → cirrhosis → jaundice, coagulopathy
│ ├─→ Brain: neurotoxicity + poor ATP → cognitive impairment, ataxia
│ ├─→ Kidney: tubular dysfunction → Fanconi syndrome
│ ├─→ Neutrophils: impaired killing → E. coli sepsis
│ ├─→ Ovaries: streak ovaries → premature ovarian failure
│ └─→ Mitochondria: cytochrome c oxidase inhibition → ATP depletion
│
├─→ Galactitol accumulates ──→ Lens: osmotic swelling → cataract
│
└─→ UDP-galactose deficit ──→ Impaired glycoprotein/glycolipid synthesis
→ CNS: defective myelin, FSH glycosylation defect
Sources
- Robbins, Cotran & Kumar Pathologic Basis of Disease (10th ed.), p. 440
- Robbins & Kumar Basic Pathology, Galactosemia section
- Henry's Clinical Diagnosis and Management by Laboratory Methods, Defects in Galactose Metabolism
- Basic Medical Biochemistry (6th ed.)
- Emery's Elements of Medical Genetics and Genomics
- Machado CM et al. (2024). Galactose-1-phosphate inhibits cytochrome c oxidase and causes mitochondrial dysfunction in classic galactosemia. Biochim Biophys Acta Mol Basis Dis. PMID 38986816
- Teixeira LF et al. (2024). Classical Hereditary galactosemia: findings in patients and animal models. Metab Brain Dis. PMID 37702899
Classical galactosemia explain biochemical reason for the clinical signs and symptoms by biochemistry book
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I now have everything I need - the Lippincott pathway diagram, detailed biochemistry from Basic Medical Biochemistry (6th ed), Harrison's, and Lippincott Illustrated Reviews. Let me compile the full answer.
Classical Galactosemia: Biochemical Basis of Clinical Signs and Symptoms
(From Biochemistry Textbooks)
The Metabolic Pathway and the Block
Galactose, derived from lactose in milk, is normally metabolized via the Leloir pathway through three sequential reactions:
(1) Galactose + ATP →[Galactokinase (GALK)]→ Galactose-1-phosphate + ADP
(2) Galactose-1-P + UDP-glucose →[GALT]→ UDP-galactose + Glucose-1-phosphate
(3) UDP-galactose →[UDP-hexose 4-epimerase]→ UDP-glucose (enters glycolysis/glycogenesis)
Net: Galactose + ATP → Glucose-1-phosphate + ADP
In classical galactosemia, step (2) is blocked due to a severe deficiency or absence of galactose-1-phosphate uridylyltransferase (GALT). The most common mutation is Q188R (homozygous), causing absent or barely detectable enzyme activity.
(Basic Medical Biochemistry 6e; Lippincott Biochemistry 8e, Chapter 12)
The Metabolic Pathway Diagram
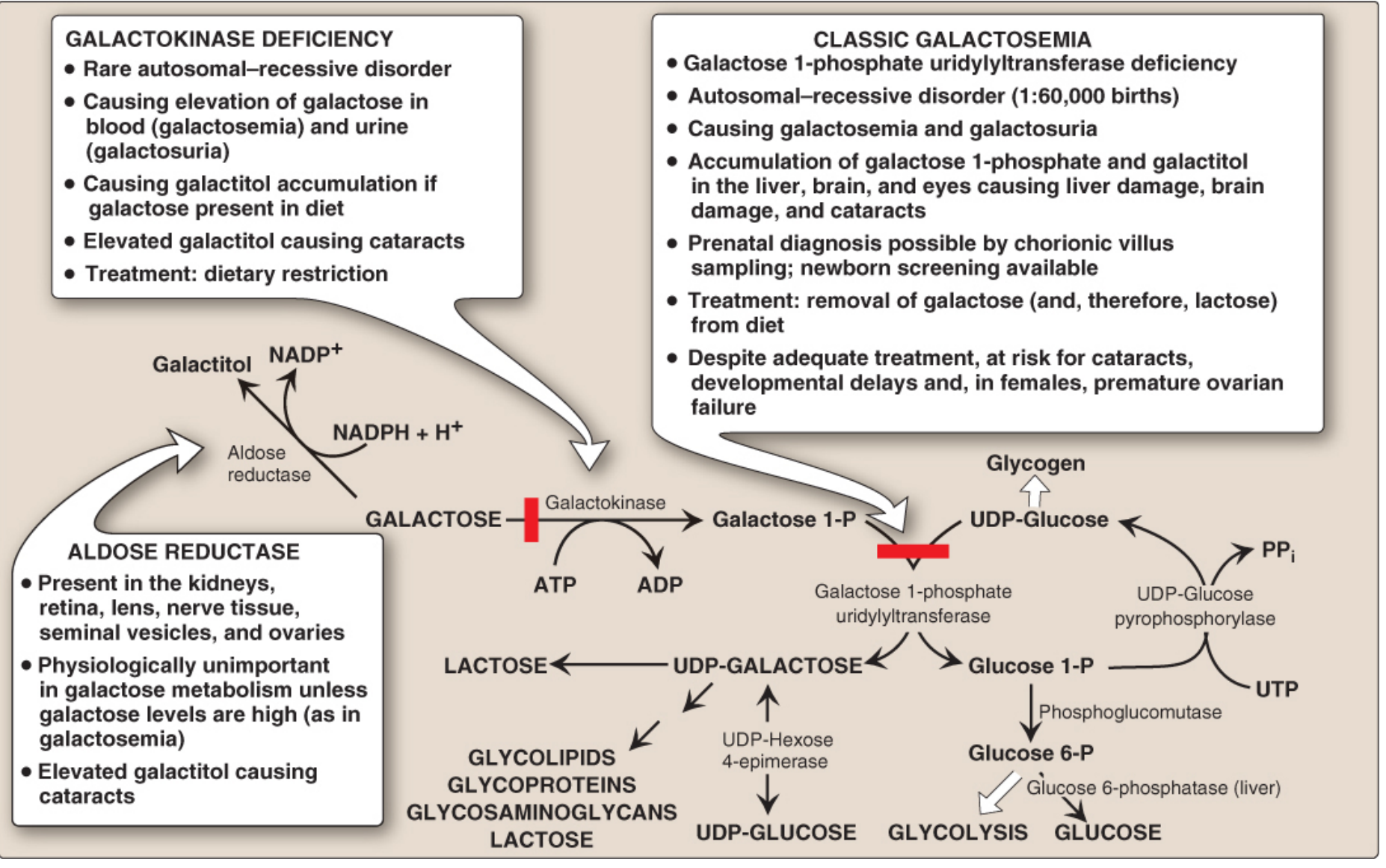
Figure 12.5, Lippincott Illustrated Reviews: Biochemistry, 8th ed. - The red bars indicate the enzymatic blocks. In classical galactosemia, GALT (right bar) is deficient, causing galactose-1-P accumulation. The aldose reductase side pathway generates galactitol, which accumulates in the lens, kidneys, and nerves.
Toxic Metabolites That Accumulate
| Metabolite | Origin | Why it accumulates |
|---|---|---|
| Galactose-1-phosphate (Gal-1-P) | Galactokinase acts normally; GALT is blocked | Primary toxic metabolite - cannot be cleared |
| Galactose | Upstream buildup from Gal-1-P backup | Elevated in blood (galactosemia) and urine (galactosuria) |
| Galactitol | Aldose reductase reduces excess galactose using NADPH | Trapped in tissues; cannot be further metabolised |
Additionally, there is a deficit of UDP-galactose - the downstream product that is essential for synthesising glycoproteins, glycolipids, glycosaminoglycans, and lactose.
(Lippincott Biochemistry 8e, p. 414-416; Basic Medical Biochemistry 6e)
Biochemical Explanation of Each Clinical Sign/Symptom
1. Failure to Thrive, Vomiting, Diarrhea (within days of first milk feed)
- Mechanism: Gal-1-P is a phosphate trap - it sequesters inorganic phosphate, depleting the cell of the phosphate needed for ATP synthesis, glycolysis, and gluconeogenesis.
- Gal-1-P also inhibits phosphoglucomutase, the enzyme that converts glucose-1-phosphate to glucose-6-phosphate, thereby impairing glycogenolysis and gluconeogenesis.
- The result is cellular energy failure in liver and intestinal cells, causing feeding intolerance. Symptoms start as soon as the infant ingests breast milk or milk-based formula - seen as early as the second day of life.
(Basic Medical Biochemistry 6e; Harrison's Internal Medicine 22e)
2. Jaundice and Hepatomegaly (first week of life)
- Early: Gal-1-P accumulation in hepatocytes causes fatty change (steatosis) by disrupting normal lipid metabolism. The liver enlarges from fat-laden cells.
- Later: Direct toxicity of Gal-1-P to hepatocytes leads to cell death, inflammation, and widespread hepatic fibrosis/cirrhosis (resembling alcoholic cirrhosis).
- Jaundice is both unconjugated (from hemolysis - see below) and conjugated (from hepatocellular damage with impaired bilirubin conjugation and excretion).
- Coagulopathy / easy bruisability: Damaged liver fails to synthesize clotting factors (factors II, VII, IX, X). Newborn screening must be done promptly because hepatic failure is rapidly progressive.
(Basic Medical Biochemistry 6e, Clinical Comments; Harrison's 22e)
3. Cataracts
This is the most precisely understood mechanism in galactosemia:
- Excess galactose in blood enters the lens of the eye (which lacks GALT and is essentially a galactose sink).
- In the lens, galactose is reduced by aldose reductase to galactitol (using NADPH as cofactor).
- Aldose reductase has a relatively high Km for galactose (~12-20 mM), so galactitol is only formed when galactose levels are substantially elevated, as in galactosemia.
- Galactitol cannot be further metabolised (there is no pathway to process it) and diffuses out of the lens very slowly - it becomes trapped.
- The accumulated galactitol creates a hyperosmotic state inside lens cells, drawing water in. The lens swells, and fiber cells rupture - producing opacification (cataract).
"Aldose reductase has a relatively high Km for galactose, approximately 12-20 mM, so galactitol is formed only in galactosemic patients who have eaten galactose. Galactitol is not further metabolized and diffuses out of the lens very slowly. Thus, hypergalactosemia is even more likely to cause cataracts than hyperglycemia."
- Basic Medical Biochemistry, 6th ed.
Early lens changes appear as "oil-drop" changes visible on slit-lamp examination, progressing to dense cataracts within days to weeks of birth.
(Basic Medical Biochemistry 6e; Lippincott Biochemistry 8e, p. 416)
4. Intellectual Disability / Brain Damage
Multiple biochemical insults converge on the brain:
| Mechanism | Detail |
|---|---|
| Gal-1-P neurotoxicity | Gal-1-P accumulates in neurons and cerebral cortex; disrupts membrane integrity and phosphate metabolism |
| UDP-galactose deficit | UDP-galactose is essential for synthesizing cerebrosides (myelin glycolipids) and glycoproteins of neuronal membranes. Deficiency impairs myelin formation and neuronal signalling - especially during the critical developmental window |
| Galactitol in brain | Galactitol accumulates in neuronal tissues, exerting osmotic damage |
| Oxidative stress | GALT deficiency is associated with increased oxidative stress and insufficient antioxidant protection - lipid peroxidation and membrane damage occur in neurons |
| Energy failure | Gal-1-P inhibition of phosphoglucomutase and downstream glycolysis reduces ATP availability in the high-energy-demand brain |
Neuropathological changes: loss of neurons, gliosis, and edema, particularly in the dentate nuclei of the cerebellum and olivary nuclei of the medulla - explaining ataxia and speech defects even in treated patients.
"One of the most serious problems of classical galactosemia is irreversible intellectual disability."
- Basic Medical Biochemistry, 6th ed.
(Basic Medical Biochemistry 6e; Harrison's 22e; Lippincott 8e)
5. Aminoaciduria / Renal Fanconi Syndrome
- Gal-1-P accumulates in renal proximal tubular cells, impairing the sodium-dependent amino acid transporters on the luminal membrane.
- The result is generalised proximal tubular dysfunction: aminoaciduria, albuminuria, glycosuria, and metabolic acidosis (renal Fanconi syndrome).
- Urine is positive for a reducing sugar (galactose) but negative for glucose on glucose-oxidase test - a key diagnostic clue. A non-glucose reducing sugar in urine of a sick neonate should immediately raise suspicion for galactosemia.
(Basic Medical Biochemistry 6e; Harrison's 22e)
6. E. coli Septicemia (Neonatal)
- Gal-1-P accumulation in neutrophils and other leukocytes impairs their bactericidal function (oxidative burst / phagocytic killing capacity).
- There is an abnormality in leukocyte function during the neonatal period, increasing susceptibility to Gram-negative sepsis, particularly Escherichia coli.
- This is especially dangerous because the onset of sepsis may actually precede the clinical diagnosis of galactosemia.
(Harrison's Internal Medicine 22e; Henry's Clinical Diagnosis)
7. Hemolysis and Coagulopathy
- Gal-1-P accumulates in red blood cells (RBCs contain GALT normally; in its absence, Gal-1-P builds up).
- RBC membrane function is disrupted, leading to hemolysis, which worsens the jaundice.
- Coagulopathy arises from hepatic failure (impaired synthesis of clotting factors).
(Basic Medical Biochemistry 6e)
8. Premature Ovarian Failure (Long-term, even on treatment)
Two biochemical mechanisms are responsible:
- UDP-galactose deficiency impairs glycoprotein synthesis: FSH and its receptor are glycoproteins. Defective glycosylation (because UDP-galactose is unavailable) impairs FSH receptor expression and signalling on ovarian follicles, disrupting oocyte maturation. This leads to hypergonadotropic hypogonadism (high FSH, low estrogen).
- Direct gonadotoxicity: Gal-1-P and galactitol accumulate in ovarian tissue, possibly impairing embryonic germ cell migration and causing early follicle depletion. "Streak ovaries" are found histologically.
Aldose reductase is expressed in the ovaries and seminal vesicles (see the pathway diagram). This is why females are predominantly affected - ovarian tissue is particularly exposed to aldose reductase-mediated galactitol accumulation. Testicular function is comparatively spared.
(Lippincott Biochemistry 8e, Fig. 12.5 clinical boxes; Harrison's 22e)
9. Hypoglycemia
- Gal-1-P inhibits phosphoglucomutase, which is needed to release glucose from glycogenolysis (glycogen → glucose-1-P → glucose-6-P → glucose).
- It also inhibits gluconeogenesis.
- Hypoglycemia is not prominent unless there is liver failure, at which point gluconeogenesis fails completely.
(Henry's Clinical Diagnosis; Harrison's 22e)
Summary Table (Biochemistry-First View)
| Clinical Feature | Primary Toxic Metabolite | Biochemical Mechanism |
|---|---|---|
| Vomiting, diarrhea, failure to thrive | Gal-1-P | Phosphate trapping; phosphoglucomutase inhibition → energy failure |
| Jaundice, hepatomegaly | Gal-1-P | Hepatocyte toxicity → steatosis → cirrhosis; impaired bilirubin processing |
| Coagulopathy, bleeding | Gal-1-P | Liver failure → reduced clotting factor synthesis |
| Cataracts | Galactitol | Aldose reductase; galactitol trapped in lens → osmotic swelling |
| Intellectual disability | Gal-1-P + UDP-Gal deficit | Neurotoxicity + impaired glycolipid/glycoprotein synthesis in CNS |
| Aminoaciduria (Fanconi) | Gal-1-P | Proximal tubule transporter inhibition |
| E. coli septicemia | Gal-1-P | Impaired neutrophil bactericidal activity |
| Hemolysis | Gal-1-P in RBCs | RBC membrane disruption |
| Premature ovarian failure | Gal-1-P + UDP-Gal deficit | Defective FSH glycosylation + direct gonadotoxicity |
| Galactosuria (reducing sugar) | Free galactose | Overflow into urine once renal threshold exceeded |
Sources
- Basic Medical Biochemistry: A Clinical Approach, 6th ed. - Chapters 22 & 27 (Galactose Metabolism; Clinical Comments, Erin G. case)
- Lippincott Illustrated Reviews: Biochemistry, 8th ed. - Chapter 12 (Fructose and Galactose Metabolism), pp. 414-416
- Harrison's Principles of Internal Medicine, 22nd ed. (2025) - Chapter 430, Galactosemia Type I
- Henry's Clinical Diagnosis and Management by Laboratory Methods - Defects in Galactose Metabolism
This is a shared conversation. Sign in to Orris to start your own chat.