Methods for detecting jaundice syndrome (hyperbilirubinemia), it and like types jaundice, and give this topics for every type Etiology ,pathology, diagnostic,mechanism, medication,normal physiology,, physical examination, therapy,related syndromes, complications
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
I now have comprehensive information from multiple authoritative textbooks. Let me compile the complete, structured response.
Jaundice (Hyperbilirubinemia) — Comprehensive Review
Normal Physiology of Bilirubin Metabolism
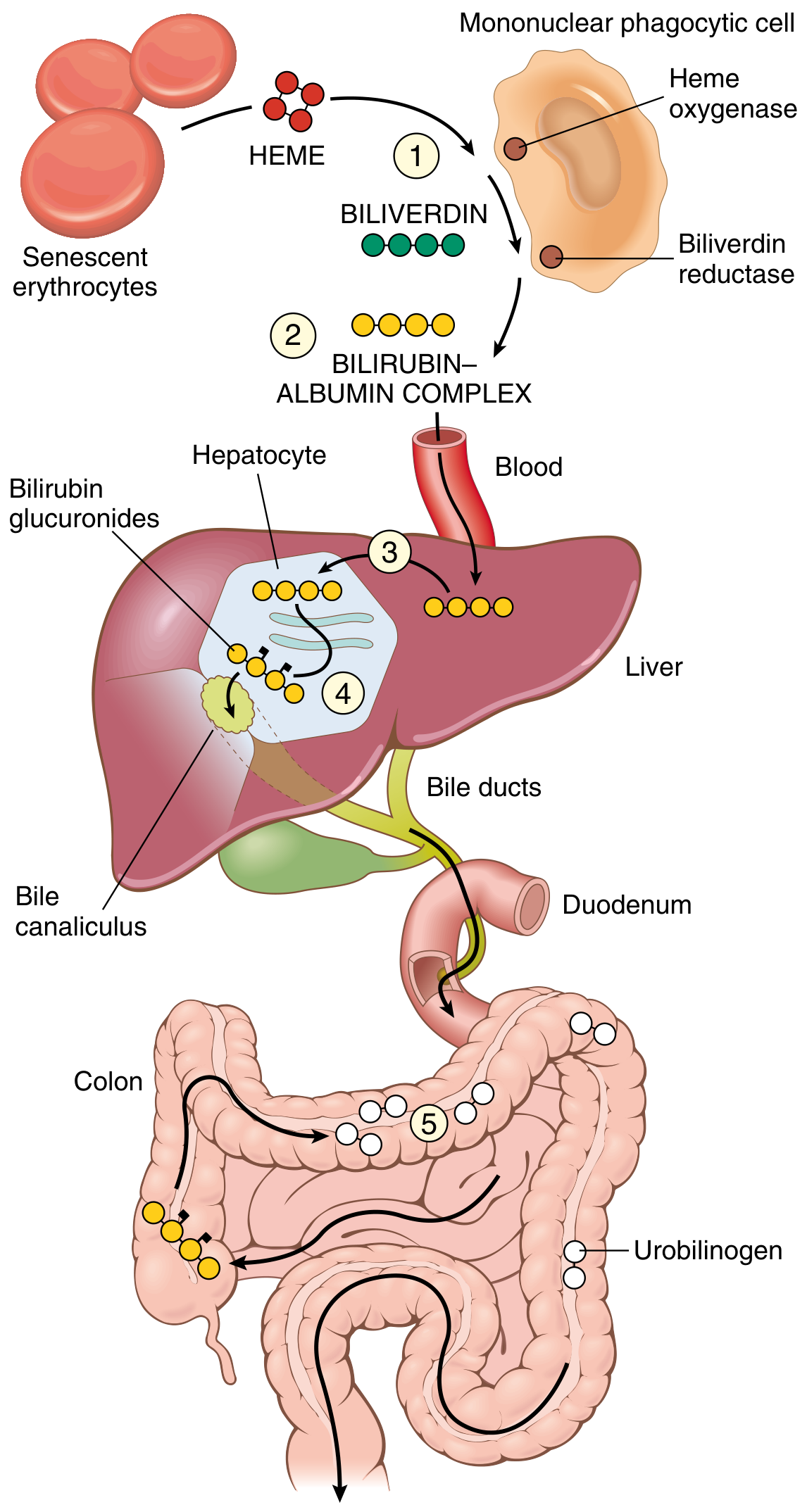
Fig. Bilirubin metabolism and elimination — Robbins, Cotran & Kumar Pathologic Basis of Disease
Bilirubin is the end product of heme catabolism. The complete pathway:
- Production (85%): Senescent red blood cells are phagocytosed by mononuclear phagocytes in the spleen, liver, and bone marrow. Heme is oxidized by heme oxygenase to biliverdin, then reduced to unconjugated (indirect) bilirubin by biliverdin reductase. The remaining 15% comes from turnover of heme-containing hepatic proteins (cytochrome P-450).
- Transport: Unconjugated bilirubin is insoluble at physiologic pH and is carried in blood bound to albumin.
- Hepatic uptake: At the sinusoidal membrane of hepatocytes, bilirubin is taken up by facilitated transport and bound to glutathione-S-transferase proteins.
- Conjugation: In the endoplasmic reticulum, UGT1A1 (UDP-glucuronosyltransferase 1A1) conjugates bilirubin with glucuronic acid, producing water-soluble bilirubin mono- and diglucuronides (conjugated/direct bilirubin).
- Canalicular excretion: Conjugated bilirubin is secreted into bile via the ATP-dependent transporter MRP2 (ABCC2). When MRP2 is overwhelmed, MRP3 secretes it back into sinusoidal blood.
- Intestinal processing: Bacteria in the ileum/colon deconjugate bilirubin and reduce it to colorless urobilinogens and stercobilin. Stercobilin is excreted in feces (brown color). ~20% of urobilinogen is reabsorbed, returned to the liver (enterohepatic circulation), and re-excreted; a small fraction escapes into urine (normal urine urobilinogen ≤4 mg/day).
Normal serum bilirubin: Total 0.2–1.2 mg/dL (indirect/unconjugated ≤1.0 mg/dL; direct/conjugated ≤0.3 mg/dL).
Jaundice threshold: Clinically visible at total bilirubin >2.5–3.0 mg/dL (scleral icterus detectable at >5 mg/dL in neonates).
— Robbins, Cotran & Kumar Pathologic Basis of Disease; Goldman-Cecil Medicine; Rosen's Emergency Medicine
Definition & Detection
Jaundice (from French jaune, "yellow") is yellow-orange discoloration of skin, sclerae, and mucous membranes due to elevated plasma bilirubin. It becomes clinically evident when total bilirubin exceeds 2.5–3 mg/dL (varies with skin pigmentation and lighting). First evident:
- Sublingually
- Conjunctival sclerae (scleral icterus)
- Hard palate
— Rosen's Emergency Medicine; Goldman-Cecil Medicine
Classification of Hyperbilirubinemia
Jaundice is classified into three major pathophysiologic categories, each with distinct subtypes:
TYPE 1 — PRE-HEPATIC (HEMOLYTIC) JAUNDICE — Unconjugated Hyperbilirubinemia
Etiology
- Hemolytic anemias (hereditary spherocytosis, sickle cell disease, G6PD deficiency, thalassemia)
- Immune-mediated hemolysis (ABO/Rh incompatibility in neonates, autoimmune hemolytic anemia)
- Mechanical destruction (prosthetic heart valves, TTP, HUS, DIC)
- Malaria
- Ineffective erythropoiesis
- Massive hematoma resorption, cephalhematoma (neonates)
Mechanism
Excess production of unconjugated bilirubin overwhelms the liver's conjugation capacity. The liver cannot process the increased bilirubin load fast enough → unconjugated bilirubin accumulates in blood. Because unconjugated bilirubin is albumin-bound, it does not spill into urine (no bilirubinuria = "acholuric jaundice").
Pathology
- Increased RBC destruction → increased heme → increased unconjugated bilirubin
- Liver histology: hemosiderin deposits, erythrophagocytosis in Kupffer cells (in chronic hemolysis)
- Splenomegaly from increased phagocytic activity
Diagnostic Tests
| Test | Finding |
|---|---|
| Total bilirubin | ↑ (predominantly indirect) |
| Direct (conjugated) bilirubin | Normal or minimally elevated |
| Urine bilirubin | Absent (unconjugated is not filtered) |
| Urine urobilinogen | Markedly ↑ |
| Fecal urobilinogen | ↑ (dark stools) |
| CBC | ↓ Hemoglobin, ↑ reticulocytes |
| LDH | ↑ |
| Haptoglobin | ↓ (binds free hemoglobin) |
| Peripheral smear | Spherocytes, schistocytes, sickled cells (depending on cause) |
| Direct Coombs test | Positive in immune hemolysis |
Physical Examination
- Jaundice (mild to moderate; rarely severe unless very brisk hemolysis)
- Pallor (anemia)
- Splenomegaly
- No hepatomegaly
- Stools and urine darkened
Therapy
- Treat underlying cause (e.g., steroids for autoimmune hemolytic anemia, folic acid, splenectomy if indicated)
- Phototherapy/exchange transfusion in neonatal hemolytic disease (if bilirubin approaches kernicterus threshold)
- Avoid triggers (e.g., oxidant drugs in G6PD deficiency)
Complications
- Kernicterus (in neonates): unconjugated bilirubin crosses blood-brain barrier → basal ganglia/brainstem deposition → athetosis, hearing loss, intellectual disability, death
- Bilirubin gallstones (pigment stones) from chronic hemolysis
- Aplastic crisis (parvovirus B19)
- High-output cardiac failure (severe anemia)
Related Syndromes
- Neonatal hemolytic disease (ABO/Rh)
- HELLP syndrome (hemolysis + elevated liver enzymes + low platelets in pregnancy)
- Sickle cell disease, thalassemia
TYPE 2 — HEPATIC (HEPATOCELLULAR) JAUNDICE — Mixed or Unconjugated Hyperbilirubinemia
This category includes inherited metabolic disorders of bilirubin handling and acquired hepatocellular disease.
2A. PHYSIOLOGIC JAUNDICE OF THE NEWBORN
Etiology / Mechanism
Transient physiologic state in the first week of life caused by:
- Decreased UGT1A1 activity in neonatal hepatocytes
- Shortened neonatal RBC lifespan → increased hemoglobin turnover
- Relative polycythemia
- Decreased intestinal bacterial colonization → decreased bilirubin conversion → increased enterohepatic recirculation
Pathology
No structural liver pathology. Purely functional immaturity.
Diagnostic Criteria
- Slow rise in bilirubin (<5 mg/dL per 24 hours)
- Peak of 5–6 mg/dL at days 2–4 of life
- Falls to <2 mg/dL by day 5–7
- Predominantly unconjugated
Physical Examination
- Jaundice progresses in a cephalocaudal direction (head → trunk → extremities)
- Scleral icterus at bilirubin >5 mg/dL
- No hepatosplenomegaly, no acholic stools
Therapy
- Phototherapy (blue-spectrum light, 390–470 nm): converts bilirubin to water-soluble photoisomers excreted in bile
- Adequate feeding (reduces enterohepatic circulation)
- Exchange transfusion for levels nearing kernicterus threshold
Complications
- Kernicterus if untreated severe neonatal hyperbilirubinemia
— Tintinalli's Emergency Medicine; Goldman-Cecil Medicine
2B. GILBERT SYNDROME
Etiology
Autosomal recessive (or dominant with variable penetrance) mutation in the UGT1A1 gene promoter region (TA repeat in TATA box: 7 TA repeats instead of normal 6), reducing UGT1A1 expression to ~30% of normal. Affects ~5–10% of the population. Benign.
Mechanism
Decreased UGT1A1 activity → mildly impaired conjugation → mild unconjugated hyperbilirubinemia. Precipitated by fasting, illness, dehydration, stress, alcohol, strenuous exercise, and menstruation.
Pathology
No structural liver abnormality.
Diagnostic Tests
- Total bilirubin: mildly elevated (1–6 mg/dL), predominantly unconjugated
- Liver enzymes (AST, ALT, ALP): normal
- CBC: normal
- Urinalysis: no bilirubinuria, urobilinogen normal
- No hemolysis markers
- Genetic testing: UGT1A1*28 promoter variant
Physical Examination
- Mild intermittent scleral icterus
- No hepatosplenomegaly
- Clinically well
Therapy
No treatment required. Reassurance. Avoid triggers.
Related Syndromes
- Crigler-Najjar syndrome (more severe UGT1A1 deficiency)
- HIV protease inhibitor-induced unconjugated hyperbilirubinemia (indinavir/atazanavir inhibit UGT1A1 via a similar mechanism)
Complications
None clinically significant. Awareness important to prevent unnecessary workup.
2C. CRIGLER-NAJJAR SYNDROME
Etiology
Rare autosomal recessive mutations in the UGT1A1 gene (coding region).
- Type I: Complete absence of UGT1A1 → severe unconjugated hyperbilirubinemia (>20 mg/dL)
- Type II (Arias syndrome): Partial deficiency → bilirubin usually 6–20 mg/dL; responds to phenobarbital
Mechanism
Complete (Type I) or near-complete (Type II) failure of bilirubin conjugation → massive unconjugated hyperbilirubinemia.
Pathology
No structural hepatic disease. Liver histology normal.
Diagnostic Tests
- Total bilirubin: markedly elevated (Type I: typically >20–25 mg/dL; Type II: 6–20 mg/dL)
- Predominantly unconjugated
- Normal liver enzymes
- Phenobarbital test: reduces bilirubin in Type II (not Type I)
- Gene sequencing: UGT1A1 mutations
Physical Examination
- Severe persistent jaundice
- Type I: neurological signs (kernicterus) if untreated
Therapy
- Type I: Phototherapy 10–16 hours/day; liver transplantation (only cure, provides functional UGT1A1)
- Type II: Phenobarbital (induces residual UGT1A1 activity); phototherapy as needed
Complications
- Type I: Kernicterus, death without transplant
- Type II: Generally survives to adulthood with treatment
2D. DUBIN-JOHNSON SYNDROME
Etiology
Autosomal recessive defect in MRP2 (ABCC2 gene) — the canalicular export pump for conjugated bilirubin. Affects canalicular secretion.
Mechanism
Conjugation is normal, but the canalicular transporter MRP2 is non-functional → conjugated bilirubin is conjugated normally but cannot be exported into bile → backs up into blood → conjugated hyperbilirubinemia.
Pathology
- Characteristic black/dark-brown pigment in hepatocytes (centrilobular distribution) on liver biopsy — a polymer of epinephrine metabolites, not bile
- Liver architecture: normal
Diagnostic Tests
- Total bilirubin: 2–5 mg/dL (conjugated/direct predominant)
- Urinalysis: bilirubin present (conjugated bilirubin is water-soluble and filtered by kidneys)
- Urobilinogen: initially decreased (early diagnosis)
- BSP (bromsulfthalein) excretion test: characteristic secondary rise at 90 minutes
- Coproporphyrin in urine: predominantly isomer I (normal: predominantly isomer III)
- Liver biopsy: black pigment deposition (grossly black liver)
Physical Examination
- Mild chronic or intermittent jaundice
- No hepatosplenomegaly
- Clinically well otherwise
Therapy
No treatment required. Benign condition. Avoid estrogen-containing oral contraceptives (may exacerbate jaundice).
Related Syndromes
- Rotor syndrome (see below)
Complications
None significant; normal life expectancy.
2E. ROTOR SYNDROME
Etiology
Autosomal recessive loss-of-function mutations in OATP1B1 and OATP1B3 (organic anion transporting polypeptide transporters) — encoded by SLCO1B1 and SLCO1B3 genes. These transporters mediate hepatocellular reuptake of conjugated bilirubin from blood.
Mechanism
Failure of hepatic re-uptake of conjugated bilirubin secreted across the hepatocyte sinusoidal membrane (by MRP3) → conjugated bilirubin accumulates in plasma.
Diagnostic Tests
- Conjugated (direct) hyperbilirubinemia
- Urine bilirubin: present
- Coproporphyrin in urine: total coproporphyrin markedly elevated; both isomers I and III elevated (distinguishes from Dubin-Johnson)
- Liver biopsy: normal (no pigment — distinguishes from Dubin-Johnson)
Physical Examination
- Mild intermittent jaundice
- No liver enlargement
Therapy
No treatment. Benign condition.
Complications
None significant.
2F. HEPATOCELLULAR JAUNDICE (Acquired — Hepatitis, Cirrhosis)
Etiology
- Viral hepatitis (HAV, HBV, HCV, HDV, HEV)
- Alcoholic hepatitis/cirrhosis
- Non-alcoholic steatohepatitis (NASH/NAFLD)
- Drug-induced liver injury (DILI): acetaminophen, isoniazid, halothane, statins, NSAIDs
- Autoimmune hepatitis
- Wilson's disease, hemochromatosis
- Acute liver failure (any cause)
Mechanism
Hepatocyte injury or death → impaired bilirubin uptake, conjugation, AND excretion → mixed (conjugated + unconjugated) hyperbilirubinemia. Damaged hepatocytes regurgitate conjugated bilirubin into sinusoidal blood.
Pathology
Varies by cause:
- Hepatitis: lobular inflammation, hepatocyte necrosis, cholestasis
- Cirrhosis: fibrosis, regenerative nodules, loss of functional hepatic mass
- Canalicular cholestasis: bile plugs in centrilobular zones
Diagnostic Tests
| Test | Finding |
|---|---|
| Total bilirubin | ↑ (mixed) |
| AST/ALT | Markedly ↑ (hepatocellular injury pattern) |
| ALP | Mildly-moderately ↑ |
| PT/INR | ↑ (synthetic dysfunction) |
| Albumin | ↓ (chronic) |
| Urine bilirubin | Present |
| Urine urobilinogen | ↑ (early); ↓ (severe disease) |
| Viral serology | HAV IgM, HBsAg, anti-HCV, etc. |
| Liver biopsy | Definitive for type and severity |
Physical Examination
- Jaundice ± scleral icterus
- Hepatomegaly (tender in acute hepatitis)
- Splenomegaly (portal hypertension in cirrhosis)
- Spider angiomata, palmar erythema, leukonychia (chronic liver disease)
- Asterixis (hepatic encephalopathy)
- Ascites, caput medusae, gynecomastia (advanced cirrhosis)
- Fetor hepaticus
Therapy
- Viral hepatitis: antivirals (entecavir/tenofovir for HBV; direct-acting antivirals for HCV)
- Alcoholic hepatitis: cessation of alcohol, corticosteroids (prednisolone 40 mg/day) if severe (Maddrey's Discriminant Function >32), pentoxifylline (second-line), N-acetylcysteine
- DILI: stop offending drug; N-acetylcysteine for acetaminophen toxicity (150 mg/kg IV over 60 min, then 50 mg/kg over 4 h, then 100 mg/kg over 16 h)
- Autoimmune hepatitis: prednisone ± azathioprine
- Acute liver failure: liver transplantation
Related Syndromes
- HELLP syndrome (pregnancy)
- Budd-Chiari syndrome (hepatic vein obstruction → congestive hepatopathy)
- Wilson's disease, hemochromatosis
Complications
- Hepatic encephalopathy
- Coagulopathy, variceal bleeding
- Hepatorenal syndrome
- Spontaneous bacterial peritonitis (SBP)
- Hepatocellular carcinoma (chronic HBV/HCV, cirrhosis)
- Acute liver failure
2G. INTRAHEPATIC CHOLESTASIS
Etiology
- Primary biliary cholangitis (PBC): autoimmune destruction of intrahepatic small bile ducts; anti-mitochondrial antibody (AMA) positive
- Primary sclerosing cholangitis (PSC): fibroinflammatory stricturing of large intra- and extrahepatic ducts; associated with IBD (ulcerative colitis in ~70%)
- Intrahepatic cholestasis of pregnancy (ICP): hormonal (estrogen/progesterone); reversible postpartum
- Drug-induced cholestasis: chlorpromazine, anabolic steroids, oral contraceptives, amoxicillin-clavulanate, rifampin
- Sepsis-associated cholestasis
- Total parenteral nutrition (TPN) cholestasis
Mechanism (PSC example)
Biliary inflammation → periductal "onion-skin" fibrosis → progressive obliteration → cholestasis → biliary cirrhosis.
Pathology (PSC)
- Concentric periductal fibrosis ("onion-skin" pattern) around atrophic ducts
- "Tombstone" scars replacing obliterated ducts
- Risk of biliary intraepithelial neoplasia → cholangiocarcinoma
Diagnostic Tests
| Test | Finding |
|---|---|
| Bilirubin | ↑ conjugated |
| ALP | Markedly ↑ (cholestatic pattern) |
| GGT | ↑ |
| AST/ALT | Mildly ↑ |
| AMA | Positive in PBC (titre >1:40) |
| pANCA | Often positive in PSC |
| ERCP/MRCP | "Beading" (alternating strictures and dilations) in PSC |
| Liver biopsy | Periductal fibrosis, ductopenia |
Physical Examination
- Jaundice
- Pruritus (bile salt deposition in skin — often severe)
- Xanthelasma/xanthomas (chronic PBC)
- Hepatomegaly
- Signs of portal hypertension (advanced disease)
- Dark urine (bilirubinuria), pale/acholic stools
Therapy
- PBC: Ursodeoxycholic acid (UDCA) 13–15 mg/kg/day (first-line); obeticholic acid (second-line); cholestyramine for pruritus; liver transplant for end-stage
- PSC: UDCA (limited evidence); ERCP with dilation/stenting of dominant strictures; liver transplant
- ICP: UDCA; early delivery (usually at 37 weeks)
- Cholestyramine/rifampicin for pruritus
- Fat-soluble vitamin supplementation (A, D, E, K)
Related Syndromes
- PSC strongly associated with ulcerative colitis
- PBC associated with Sjögren's syndrome, CREST syndrome
- IgG4-related sclerosing cholangitis (steroid-responsive; mimics PSC)
Complications
- Biliary cirrhosis → portal hypertension
- Cholangiocarcinoma (PSC — lifetime risk ~10–15%)
- Fat malabsorption, steatorrhea, osteoporosis (fat-soluble vitamin deficiency)
- Hepatocellular carcinoma (end-stage)
TYPE 3 — POST-HEPATIC (OBSTRUCTIVE/CHOLESTATIC) JAUNDICE — Conjugated Hyperbilirubinemia
Etiology
Intraluminal obstruction:
- Choledocholithiasis (common bile duct stones)
- Biliary stricture (post-surgical, post-inflammatory)
- Parasites (Ascaris, Clonorchis)
Mural obstruction:
- Cholangiocarcinoma
- Primary sclerosing cholangitis
Extraluminal obstruction:
- Carcinoma of head of pancreas (classic: painless progressive jaundice)
- Ampullary carcinoma
- Periportal lymphadenopathy
- Pancreatitis (inflammatory mass)
- Mirizzi syndrome (gallstone in cystic duct compressing CBD)
Mechanism
Obstruction of the common bile duct or intrahepatic ducts → bile cannot drain into duodenum → conjugated bilirubin backs up into hepatocytes → regurgitates into sinusoidal blood → conjugated hyperbilirubinemia → bilirubinuria → acholic stools (no bilirubin in gut).
Pathology
- Bile duct dilatation proximal to obstruction (detectable on ultrasound: CBD >6 mm)
- Canalicular bile plugs
- Bile infarcts (focal hepatocellular necrosis from bile salt toxicity)
- Portal tract edema and neutrophilic infiltrate (cholangitis)
- Longstanding obstruction: secondary biliary cirrhosis
Diagnostic Tests
| Test | Finding |
|---|---|
| Total bilirubin | ↑ predominantly conjugated (direct) |
| ALP | Markedly ↑ (>3× ULN) |
| GGT | ↑ |
| AST/ALT | Mildly ↑ |
| Urine bilirubin | Strongly positive (bilirubinuria) |
| Urine urobilinogen | Absent/decreased (no bilirubin reaching gut) |
| Stool color | Pale/clay-colored (acholic) |
| CA 19-9 | ↑ in pancreatic/cholangiocarcinoma |
| Ultrasound (first-line) | Dilated bile ducts; gallstones |
| CT abdomen | Mass lesion, pancreatic head tumor |
| MRCP/ERCP | Defines level and cause of obstruction |
| Endoscopic ultrasound (EUS) | Fine-needle aspiration of mass |
Imaging priority: Ultrasound is the preferred initial modality; CT preferred if malignancy suspected. — Rosen's Emergency Medicine
Physical Examination
- Progressive jaundice (often painless if malignant; colicky pain if calculous)
- Pruritus (bile salts in skin)
- Dark urine ("Coca-Cola" urine)
- Acholic (clay-colored/pale) stools
- Courvoisier's sign: palpable, non-tender gallbladder in painless obstructive jaundice → suggests malignant obstruction (pancreatic head carcinoma), not calculous disease (where gallbladder is scarred)
- Tenderness in right upper quadrant (Charcot's triad in cholangitis: RUQ pain + fever + jaundice)
- Hepatomegaly (congestion of bile)
- Signs of sepsis if ascending cholangitis present (Reynolds' pentad: Charcot's triad + hypotension + confusion)
Therapy
Calculous obstruction:
- ERCP with sphincterotomy and stone extraction
- Laparoscopic cholecystectomy after CBD clearance
- Antibiotics for cholangitis (piperacillin-tazobactam or cefotaxime + metronidazole)
Malignant obstruction:
- Resectable pancreatic head cancer: Whipple procedure (pancreaticoduodenectomy) ± adjuvant gemcitabine/capecitabine
- Unresectable: palliative ERCP biliary stenting or percutaneous transhepatic cholangiography (PTC)
- Cholangiocarcinoma: surgical resection (if possible); biliary drainage
Biliary stricture:
- Endoscopic or surgical repair; balloon dilation and stenting
Related Syndromes
- Charcot's triad (cholangitis): RUQ pain, fever, jaundice
- Reynolds' pentad: adds shock and mental status change (suppurative cholangitis)
- Mirizzi syndrome: external compression of CBD by a large gallstone in the cystic duct
- Courvoisier's law: painless jaundice + palpable gallbladder = malignant obstruction (not stones)
Complications
- Ascending cholangitis (bacterial infection proximal to obstruction) → sepsis, liver abscesses
- Secondary biliary cirrhosis (chronic obstruction)
- Hepatorenal syndrome
- Coagulopathy (vitamin K malabsorption → factor II, VII, IX, X deficiency)
- Malnutrition/steatorrhea (no bile in gut)
Summary Table: Differential Features of Jaundice Types
| Feature | Pre-hepatic (Hemolytic) | Hepatocellular | Obstructive (Post-hepatic) |
|---|---|---|---|
| Bilirubin type | Unconjugated ↑↑ | Mixed ↑ | Conjugated ↑↑ |
| Urine bilirubin | Absent | Present | Strongly present |
| Urine urobilinogen | ↑↑ | ↑ early / ↓ severe | Absent |
| Stool color | Dark (↑ stercobilin) | Variable | Pale/acholic |
| ALP | Normal | Mild ↑ | Markedly ↑ |
| AST/ALT | Normal | Markedly ↑ | Mild ↑ |
| Haptoglobin | ↓ | Normal/↓ | Normal |
| Imaging | Normal | Hepatomegaly | Dilated ducts |
| Courvoisier sign | Negative | Negative | Positive (malignant) |
Medications Causing Jaundice (Drug-Induced)
| Drug | Mechanism |
|---|---|
| Acetaminophen | Hepatocellular necrosis (zone 3) via NAPQI toxic metabolite |
| Isoniazid, rifampin | Hepatocellular DILI |
| Statins | Hepatocellular DILI (rare) |
| Chlorpromazine, anabolic steroids | Cholestatic DILI |
| Amoxicillin-clavulanate | Mixed/cholestatic DILI |
| Atazanavir, indinavir (HIV PI) | UGT1A1 inhibition → unconjugated hyperbilirubinemia (Gilbert-like) |
| Estrogens/OCPs | Intrahepatic cholestasis |
| Rifampin | Inhibits OATP1B1/1B3 uptake transporters |
Related Syndromes — Quick Reference
| Syndrome | Key Feature |
|---|---|
| Gilbert syndrome | Benign UGT1A1 promoter variant; triggers: fasting, illness |
| Crigler-Najjar I | Complete UGT1A1 absence; kernicterus without transplant |
| Crigler-Najjar II (Arias) | Partial deficiency; responds to phenobarbital |
| Dubin-Johnson | MRP2 defect; black liver on biopsy; conjugated hyperbilirubinemia |
| Rotor syndrome | OATP1B1/B3 defect; conjugated hyperbilirubinemia; normal biopsy |
| PBC | AMA+; intrahepatic duct destruction; middle-aged women |
| PSC | "Beading" on ERCP/MRCP; associated with UC; risk of cholangiocarcinoma |
| ICP | Pregnancy; UDCA treatment; early delivery |
| HELLP | Hemolysis + elevated LFTs + low platelets in pregnancy |
| Budd-Chiari | Hepatic vein thrombosis; congestive hepatopathy; jaundice + ascites + RUQ pain |
| Kernicterus | Unconjugated bilirubin crossing blood-brain barrier in neonates → basal ganglia injury |
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease; Goldman-Cecil Medicine; Rosen's Emergency Medicine; Tintinalli's Emergency Medicine; Harper's Illustrated Biochemistry, 32nd Ed.; Sleisenger & Fordtran's Gastrointestinal and Liver Disease
Can u give ippa for all types
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive textbook content for a detailed IPPA (Inspection, Palpation, Percussion, Auscultation) breakdown for all jaundice types. Here is the complete structured answer:
IPPA Examination in Jaundice (Hyperbilirubinemia) — All Types
IPPA = Inspection → Palpation → Percussion → Auscultation
General IPPA Sequence for Any Jaundiced Patient
Before type-specific findings, every jaundiced patient must be examined in sequence:
Start with the conjunctivae, sublingual mucosa, and hard palate — the earliest sites of visible jaundice (bilirubin >2.5 mg/dL). Progression is cephalocaudal (face → trunk → extremities), though the degree of spread does NOT reliably correlate with exact bilirubin level.
— Rosen's Emergency Medicine; Tintinalli's Emergency Medicine
TYPE 1 — PRE-HEPATIC JAUNDICE (Hemolytic)
INSPECTION
| Region | Finding | Significance |
|---|---|---|
| Sclerae / conjunctivae | Mild scleral icterus | Bilirubin elevation, usually mild |
| Skin | Mild yellow tint; pallor prominent | Anemia from hemolysis |
| Skin color | Lemon-yellow (pale yellow) tinge | Combination of jaundice + anemia |
| Urine | Dark amber / "Coca-Cola" colored | ↑ urobilinogen (not bilirubinuria) |
| Stool | Dark brown / very dark | ↑ stercobilin from excess bilirubin load |
| Scleral icterus | Present but mild | Unconjugated bilirubin rarely exceeds 4–5 mg/dL in chronic hemolysis |
| Face / pallor | Pale conjunctivae, pale mucous membranes | Hemolytic anemia |
| Jaundice extent | Usually mild; no acholic stools | Conjugation and excretion are intact |
PALPATION
| Region | Finding | Significance |
|---|---|---|
| Liver | Not enlarged (normal in most) OR mildly enlarged if hemosiderin deposition | No primary hepatic disease |
| Spleen | Splenomegaly (often significant) | Increased phagocytic destruction of RBCs |
| Lymph nodes | Non-tender; may be enlarged in lymphoma-associated hemolysis | Rule out malignant cause |
| Gallbladder | Non-palpable (normal) | No obstruction |
| Abdomen | Soft, non-tender (unless splenic infarct in sickle cell — acute left-sided pain and tenderness) |
PERCUSSION
| Region | Finding | Significance |
|---|---|---|
| Liver span | Normal to slightly increased (hepatic dullness) | Liver not primarily affected |
| Spleen | Increased splenic dullness extending beyond normal limits | Splenomegaly confirmed |
| Traube's space (left lower anterior chest) | Dull (instead of resonant) | Splenic enlargement |
| Abdomen general | Resonant; no shifting dullness | No ascites in uncomplicated hemolysis |
AUSCULTATION
| Region | Finding | Significance |
|---|---|---|
| Abdomen | Normal bowel sounds | No obstruction |
| Heart | May have flow murmur (systolic, ejection type) | High-output state from anemia |
| No bruits | Absent hepatic or splenic bruits | No portal hypertension |
TYPE 2A — PHYSIOLOGIC JAUNDICE OF THE NEWBORN
INSPECTION
| Region | Finding | Significance |
|---|---|---|
| Skin | Yellow discoloration beginning at face, progressing to trunk and extremities (cephalocaudal) | Normal physiologic pattern |
| Sclerae | Scleral icterus at bilirubin >5 mg/dL | Earliest visible sign |
| Stool | Normal yellow color (transitional stools) | Conjugation and excretion intact |
| Urine | Normal color (no bilirubinuria) | Unconjugated bilirubin, not renally excreted |
| Fontanelle | Flat and soft (normal) | No kernicterus-associated increased ICP |
| Alertness | Active, feeding well | Physiologic state |
| Head | No cephalhematoma (if present → suspect pathologic) | Hematoma → ↑ bilirubin load |
Blanch test: Press skin on nose/forehead — yellow color appears = jaundice confirmed clinically.
PALPATION
| Region | Finding | Significance |
|---|---|---|
| Liver | Not enlarged (normal) | No hepatic disease |
| Spleen | Not enlarged | No hemolytic disease |
| Abdomen | Soft, non-tender | Normal |
| Fontanelle | Flat | Normal; bulging fontanelle → meningitis/kernicterus |
PERCUSSION
| Region | Finding | Significance |
|---|---|---|
| Liver | Normal span for neonate (4–5 cm) | No hepatomegaly |
| Abdomen | Resonant throughout | No organomegaly |
AUSCULTATION
| Region | Finding | Significance |
|---|---|---|
| Abdomen | Normal bowel sounds | Normal gut motility |
| Cardiac | Normal heart sounds; no murmur | No hemolytic anemia |
TYPE 2B — GILBERT SYNDROME
INSPECTION
| Region | Finding | Significance |
|---|---|---|
| Sclerae | Mild, intermittent scleral icterus — most prominent when fasting/unwell | UGT1A1 reduction — mild unconjugated ↑ |
| Skin | Mild yellow tinge (intermittent) | Never deeply jaundiced |
| Urine | Normal color (no bilirubinuria) | Unconjugated bilirubin not renally filtered |
| Stool | Normal color | Excretion not impaired |
| General | Well-appearing, healthy | Benign condition |
PALPATION
| Region | Finding | Significance |
|---|---|---|
| Liver | Normal size, non-tender | No structural liver disease |
| Spleen | Normal | No hemolysis |
| Abdomen | Soft, non-tender | Completely normal |
PERCUSSION
| Region | Finding | Significance |
|---|---|---|
| Liver | Normal dullness, normal span | No hepatomegaly |
| Spleen | Normal | No splenomegaly |
| Abdomen | Normal resonance | No ascites |
AUSCULTATION
| Region | Finding | Significance |
|---|---|---|
| Abdomen | Normal bowel sounds | Normal |
TYPE 2C — CRIGLER-NAJJAR SYNDROME
INSPECTION
| Region | Finding | Significance |
|---|---|---|
| Skin | Deep, persistent jaundice from birth | Severe unconjugated hyperbilirubinemia |
| Sclerae | Markedly icteric | Bilirubin typically >20 mg/dL (Type I) |
| Neurologic appearance | Opisthotonus (backward arching), dystonia, hypotonia or hypertonia | Kernicterus in untreated Type I |
| Eyes | Abnormal ocular movements (oculomotor palsies) | Brainstem involvement in kernicterus |
| Alertness | Reduced consciousness, seizures in advanced kernicterus | |
| Urine | Normal (unconjugated — not renally excreted) | |
| Stool | Normal color | Excretion pathway intact |
| Hearing | Sensorineural hearing loss (Kernicterus — cochlear nuclei damage) | Clinical inspection finding |
PALPATION
| Region | Finding | Significance |
|---|---|---|
| Liver | Normal size and texture | No structural liver disease |
| Spleen | Normal | No hemolysis |
| Neurologic tone | Increased (hypertonia) or decreased (hypotonia) | Kernicterus |
PERCUSSION
| Region | Finding | Significance |
|---|---|---|
| Liver | Normal span | Normal architecture |
| Abdomen | Resonant; no ascites | No liver disease |
AUSCULTATION
| Region | Finding | Significance |
|---|---|---|
| Abdomen | Normal | Normal |
TYPE 2D — DUBIN-JOHNSON SYNDROME
INSPECTION
| Region | Finding | Significance |
|---|---|---|
| Skin | Mild-to-moderate chronic jaundice | Conjugated hyperbilirubinemia |
| Sclerae | Mild persistent icterus | Conjugated bilirubin elevated |
| Urine | Dark (brownish/tea-colored) — bilirubinuria | Conjugated bilirubin is water-soluble and renally excreted |
| Stool | Normal color | Excretion of some bilirubin still occurs |
| General appearance | Well-looking; no signs of chronic liver disease | Benign |
PALPATION
| Region | Finding | Significance |
|---|---|---|
| Liver | Normal or mildly enlarged, non-tender | Benign; may have mild hepatomegaly |
| Spleen | Normal | |
| Gallbladder | Non-palpable | Not obstructed |
PERCUSSION
| Region | Finding | Significance |
|---|---|---|
| Liver | Normal or mildly enlarged span | |
| Abdomen | No shifting dullness | No ascites |
AUSCULTATION
| Region | Finding | Significance |
|---|---|---|
| Abdomen | Normal bowel sounds | Normal |
TYPE 2E — ROTOR SYNDROME
INSPECTION
| Region | Finding | Significance |
|---|---|---|
| Skin / Sclerae | Mild conjugated jaundice | OATP1B1/1B3 defect |
| Urine | Dark (bilirubinuria) | Conjugated bilirubin excreted renally |
| Stool | Normal | |
| General | Healthy appearance; no stigmata of liver disease | Benign |
PALPATION
| Region | Finding | Significance |
|---|---|---|
| Liver | Normal | No structural pathology (biopsy is normal — distinguishes from Dubin-Johnson) |
| Spleen | Normal |
PERCUSSION / AUSCULTATION
Normal in both. No organomegaly, no ascites, no bruits.
TYPE 2F — HEPATOCELLULAR JAUNDICE (Hepatitis/Cirrhosis)
INSPECTION
| Region | Finding | Significance |
|---|---|---|
| Skin — jaundice | Moderate-to-deep jaundice | Mixed (conjugated + unconjugated) hyperbilirubinemia |
| Spider angiomata | Dilated central arteriole with radiating vessels on upper trunk, face, arms (>5 = significant) | Hyperestrogenism from impaired hepatic estrogen metabolism |
| Palmar erythema | Erythema over thenar/hypothenar eminences | Chronic liver disease |
| Leukonychia | White nails (Terry's nails) | Hypoalbuminemia |
| Dupuytren's contracture | Fibrosis of palmar fascia | Alcoholic liver disease |
| Muscle wasting | Temporal, limb atrophy | Sarcopenia of chronic liver disease |
| Caput medusae | Dilated periumbilical veins on abdomen | Portal hypertension — collateral circulation |
| Gynecomastia | Breast tissue enlargement in males | ↓ hepatic estrogen clearance |
| Xanthelasma/xanthoma | Yellow plaques near eyelids/tendons | Cholestatic liver disease (PBC) |
| Urine | Dark (bilirubinuria) | Conjugated bilirubin excreted |
| Stool | Variable: pale (cholestasis) or normal | Depends on degree of cholestasis |
| Fetor hepaticus | Sweet/musty odor of breath | ↑ dimethyl sulfide from portosystemic shunting |
| Petechiae / ecchymosis | Spontaneous bruising | Coagulopathy (↓ clotting factor synthesis) |
| Edema | Peripheral pitting edema (bilateral) | Hypoalbuminemia + sodium retention |
| Mental status | Confusion, asterixis (flapping tremor) | Hepatic encephalopathy |
Asterixis test: Ask patient to extend wrists with fingers spread — bilateral flapping tremor = positive → hepatic encephalopathy.
PALPATION
| Region | Finding | Significance |
|---|---|---|
| Liver — Acute hepatitis | Enlarged, smooth, tender hepatomegaly | Hepatocyte swelling, inflammation |
| Liver — Cirrhosis | Firm, nodular, may be shrunken — or sometimes normal size | Fibrosis/regenerative nodules; eventually atrophies |
| Splenomegaly | Enlargement to left hypochondrium | Portal hypertension (>10 mmHg) |
| Abdomen | Diffuse tenderness in acute hepatitis | Hepatic capsule stretch/inflammation |
| Fluid wave / shifting dullness | Present if ascites | Portal hypertension + hypoalbuminemia + ↑ aldosterone |
| Gallbladder | Not palpable | Collapsed due to hepatocellular disease |
PERCUSSION
| Region | Finding | Significance |
|---|---|---|
| Liver span | Increased (>12 cm) in hepatitis | Hepatomegaly |
| Liver span | Decreased (<6 cm) in cirrhosis | Hepatic atrophy/fibrosis |
| Shifting dullness | Positive (dullness shifts with position) | Ascites ≥1.5 L |
| Fluid thrill (puddle sign) | Transmitted vibration across abdomen | Large ascites |
| Splenic dullness | Extended beyond Traube's space | Splenomegaly from portal hypertension |
AUSCULTATION
| Region | Finding | Significance |
|---|---|---|
| Hepatic bruit | High-pitched systolic bruit over liver | Hepatocellular carcinoma (hypervascular), alcoholic hepatitis |
| Venous hum | Continuous hum at periumbilical area (Cruveilhier-Baumgarten murmur) | Portal hypertension with recanalized umbilical vein |
| Bowel sounds | Decreased (if ascites or paralytic ileus) | Ascites |
| Hepatic rub | Friction rub over liver (rare) | Liver infarct, abscess, malignancy |
TYPE 2G — INTRAHEPATIC CHOLESTASIS (PBC / PSC)
INSPECTION
| Region | Finding | Significance |
|---|---|---|
| Skin jaundice | Deep yellow/greenish-yellow tinge | Prolonged conjugated hyperbilirubinemia |
| Skin excoriations | Scratch marks all over body | Severe pruritus from bile salt deposition |
| Xanthelasma | Yellow plaques near medial canthi | Hypercholesterolemia (impaired bile secretion) |
| Xanthomas | Yellow nodules on tendons, extensor surfaces | Chronic cholestasis |
| Urine | Dark (bilirubinuria) | Conjugated hyperbilirubinemia |
| Stool | Pale/clay-colored (acholic) | No bilirubin reaching gut |
| Skin pigmentation | Hyperpigmentation (bronze/tan skin) | Melanin ↑ from bile salt stimulation of melanocytes |
| Eyes | Kayser-Fleischer rings (in Wilson's disease) | Copper deposition |
| Signs of IBD | Perianal skin tags, fistulas | PSC associated with ulcerative colitis in ~70% |
PALPATION
| Region | Finding | Significance |
|---|---|---|
| Liver | Enlarged, smooth, firm hepatomegaly | Intrahepatic bile accumulation/fibrosis |
| Spleen | Enlarged (portal hypertension in advanced disease) | Secondary biliary cirrhosis |
| Gallbladder | Non-palpable (small duct disease) | Not obstructed at extrahepatic level |
| Abdomen | Mild tenderness in RUQ | Hepatic capsule distension |
| Ascites | Present in advanced cirrhotic stage | Portal hypertension |
PERCUSSION
| Region | Finding | Significance |
|---|---|---|
| Liver span | Increased (hepatomegaly) | Cholestatic hepatomegaly |
| Shifting dullness | Positive in advanced disease | Ascites from biliary cirrhosis |
| Spleen | Extended dullness (Traube's space) | Splenomegaly |
AUSCULTATION
| Region | Finding | Significance |
|---|---|---|
| Abdomen | Normal or decreased bowel sounds | Ascites effect on bowel |
| No hepatic bruit | Absent | Not vascular lesion |
TYPE 3 — POST-HEPATIC OBSTRUCTIVE JAUNDICE
INSPECTION
| Region | Finding | Significance |
|---|---|---|
| Skin jaundice | Deep, progressive, greenish-yellow | High conjugated bilirubin (prolonged = bile staining) |
| Urine | Dark (tea-colored) — strong bilirubinuria | Conjugated bilirubin renally excreted |
| Stool | Pale/clay-colored/acholic (putty stools) | No bilirubin reaching intestines — pathognomonic of complete obstruction |
| Skin excoriations | Scratch marks from pruritus | Bile salt deposition in skin |
| Weight loss | Visible cachexia | Pancreatic or biliary malignancy |
| Scleral icterus | Deep icterus | High bilirubin (often >10 mg/dL in malignant obstruction) |
| Skin pigmentation | Yellow-green tinge (prolonged obstruction) | Biliverdin accumulation |
| Signs of coagulopathy | Bruising, petechiae | Vitamin K malabsorption (fat-soluble vitamin deficiency) |
| Visible mass | Epigastric or RUQ fullness | Pancreatic head mass or dilated gallbladder |
PALPATION
| Region | Finding | Significance |
|---|---|---|
| Courvoisier's sign | Palpable, smooth, NON-TENDER enlarged gallbladder | Classic: malignant obstruction (pancreatic head Ca, cholangiocarcinoma, ampullary Ca) — gallbladder not fibrosed, able to distend |
| Liver | Enlarged, smooth, non-tender | Bile congestion proximal to obstruction |
| Gallbladder | Tender, not palpable in calculous obstruction | Scarred, fibrosed gallbladder from chronic cholecystitis — cannot distend |
| Epigastric mass | Hard, irregular palpable mass | Pancreatic head carcinoma (late sign) |
| RUQ tenderness | Present in cholangitis/cholecystitis | Infection/inflammation overlaid on obstruction |
| Murphy's sign | Positive: inspiratory arrest on deep RUQ palpation | Acute cholecystitis (gallstone disease) |
| Spleen | Usually normal (unless secondary biliary cirrhosis) | |
| Ascites | Late sign | Advanced malignancy / biliary cirrhosis |
Courvoisier's Law: Painless jaundice + palpable gallbladder = malignant obstruction, NOT calculous disease. — Schwartz's Principles of Surgery
PERCUSSION
| Region | Finding | Significance |
|---|---|---|
| Liver | Increased span | Congestive hepatomegaly |
| RUQ | Dull | Enlarged liver and/or gallbladder |
| Murphy's percussion sign | Tenderness on percussion over gallbladder area | Cholecystitis |
| Abdomen | Resonant centrally | No ascites (unless advanced) |
| Shifting dullness | Present in late-stage biliary cirrhosis or malignancy | Ascites |
AUSCULTATION
| Region | Finding | Significance |
|---|---|---|
| Bowel sounds | Normal to hyperactive (if partial obstruction) | |
| Bowel sounds | Absent (if complete bowel obstruction) | Emergency |
| No hepatic bruit | Absent (unless underlying HCC) | |
| No venous hum | Absent (no portal hypertension unless biliary cirrhosis) |
Summary IPPA Comparison Table
| Feature | Pre-hepatic (Hemolytic) | Hepatocellular | Obstructive |
|---|---|---|---|
| Inspection: Jaundice | Mild, lemon-yellow | Moderate-deep, yellow | Deep, greenish-yellow |
| Inspection: Urine | Dark (↑ urobilinogen) | Dark (bilirubin + urobilinogen) | Dark (bilirubin only) |
| Inspection: Stool | Dark (↑ stercobilin) | Variable | Acholic/pale |
| Inspection: Skin signs | Pallor | Spider angiomata, caput medusae, excoriations | Excoriations, xanthomas |
| Palpation: Liver | Normal | Tender enlarged (acute) / nodular small (cirrhosis) | Smooth enlarged (congested) |
| Palpation: Spleen | Enlarged | Enlarged (portal HTN) | Normal (unless late) |
| Palpation: Gallbladder | Not palpable | Not palpable | Palpable, non-tender (Courvoisier) |
| Percussion: Liver span | Normal | ↑ (hepatitis) / ↓ (cirrhosis) | ↑ (congestion) |
| Percussion: Ascites | Absent | Present (cirrhosis) | Present (late/malignancy) |
| Auscultation: Bruit | Absent | + Hepatic bruit (HCC/AH) | Absent |
| Auscultation: Venous hum | Absent | + Cruveilhier-Baumgarten (portal HTN) | Absent |
| Murphy's sign | Negative | Negative | Positive (cholecystitis) |
| Courvoisier's sign | Negative | Negative | Positive (malignant) |
| Asterixis | Absent | Present (encephalopathy) | Absent |
— Rosen's Emergency Medicine; Tintinalli's Emergency Medicine; Schwartz's Principles of Surgery; Robbins, Cotran & Kumar Pathologic Basis of Disease
This is a shared conversation. Sign in to Orris to start your own chat.