Cardiomyopathy types
Reading File
Finding Sources
Searching the Web
cardiomyopathy types classification 2024
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Finding Sources
I now have all the material needed for a thorough, well-sourced answer. Let me compose it.
Cardiomyopathy Types
Cardiomyopathy refers to intrinsic heart muscle diseases that cause mechanical and/or electrical dysfunction, independent of coronary artery disease, valvular disease, congenital heart disease, or hypertension. There are three classic pathophysiologic categories plus several important additional subtypes.
Classification Overview
| Type | Dominant Dysfunction | Prevalence |
|---|---|---|
| Dilated (DCM) | Systolic (contractile) | ~90% of cardiomyopathies |
| Hypertrophic (HCM) | Diastolic (relaxation) | 1 in 500 general population |
| Restrictive (RCM) | Diastolic (compliance) | Least common |
| Arrhythmogenic RV (ARVC) | Electrical + RV structural | Hereditary |
| Takotsubo (Stress) | Transient systolic | Acquired |
| Peripartum | Systolic | Pregnancy-related |
| Left Ventricular Non-Compaction (LVNC) | Mixed | Congenital/genetic |
1. Dilated Cardiomyopathy (DCM)
Pathophysiology: Systolic (contractile) dysfunction with a dilated, flabby, hypocontracting ventricle. EF is reduced.
Causes:
- Genetic: Autosomal dominant loss-of-function mutations (up to 50% of cases) affecting cytoskeletal, sarcolemmal, or nuclear envelope proteins. Titin-truncation mutations account for up to 20% of DCM cases.
- Myocarditis (viral - most common infectious cause in the US)
- Toxic: alcohol, anthracyclines, cocaine
- Pregnancy (peripartum cardiomyopathy)
- Idiopathic (remaining cases)
Clinical features: Bilateral heart failure symptoms, S3 gallop, mitral regurgitation from annular dilation, risk of ventricular arrhythmia, thromboembolic events.
Disease phases:
- Phase 1A: Genetic variant present, no structural disease yet
- Phase 1B: DCM present but asymptomatic (may last years)
- Phase 2: Symptomatic heart failure, arrhythmia, or embolus
(Braunwald's Heart Disease; Robbins & Cotran Pathologic Basis of Disease)
2. Hypertrophic Cardiomyopathy (HCM)
Pathophysiology: Diastolic dysfunction from a thick-walled, heavy, hypercontracting ventricle with poor compliance. Systolic function is usually preserved.
Genetics: Virtually all cases are autosomal dominant gain-of-function mutations in sarcomeric proteins. Over 400 mutations across 9 genes are known, most commonly:
- Myosin-binding protein C (MYBP-C)
- Beta-myosin heavy chain (MYH7)
- Cardiac troponins I, T, and alpha-tropomyosin Together these account for ~70% of cases.
Key features:
- Asymmetric septal hypertrophy (septum >> free wall)
- LV outflow tract obstruction in ~1/3 of cases via systolic anterior motion (SAM) of the mitral valve
- Obstruction is dynamic: worsens with reduced preload, reduced afterload, or increased contractility
Clinical significance: Most common cardiovascular cause of sudden cardiac death in young athletes, accounting for ~1/3 of such events. Risk of SCD increases at ~1% per year.
Distinguishing from: Amyloidosis, Fabry disease, hypertensive heart disease, valvular/subvalvular aortic stenosis.
(Robbins & Cotran; Braunwald's Heart Disease; Tintinalli's Emergency Medicine)
3. Restrictive Cardiomyopathy (RCM)
Pathophysiology: Stiff, noncompliant myocardium with impaired diastolic filling. Ventricular cavity size is normal or reduced, walls may be thickened, and systolic function is often preserved early on.
Causes:
| Mechanism | Examples |
|---|---|
| Infiltration | Amyloidosis (AL, ATTR), Sarcoidosis, Gaucher disease |
| Iron deposition | Hemochromatosis |
| Storage diseases | Fabry disease, glycogen storage disorders |
| Fibrosis | Radiation-induced interstitial fibrosis |
| Endomyocardial | Endomyocardial fibrosis, Löffler eosinophilic endocarditis |
| Endocrine | Carcinoid heart disease |
Key clinical distinction: Must be differentiated from constrictive pericarditis (both cause diastolic failure but management differs radically).
Note: Amyloidosis causes restriction only in late stages when extensive infiltration of the myocardial interstitium has occurred. 4% of African Americans carry a specific transthyretin (ATTR) mutation that raises cardiac amyloidosis risk more than fourfold.
(Braunwald's Heart Disease; Goldman-Cecil Medicine)
4. Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC)
Pathophysiology: Progressive fibro-fatty replacement of the right ventricular myocardium, causing right-sided heart failure and ventricular arrhythmias. Left-sided involvement can also occur.
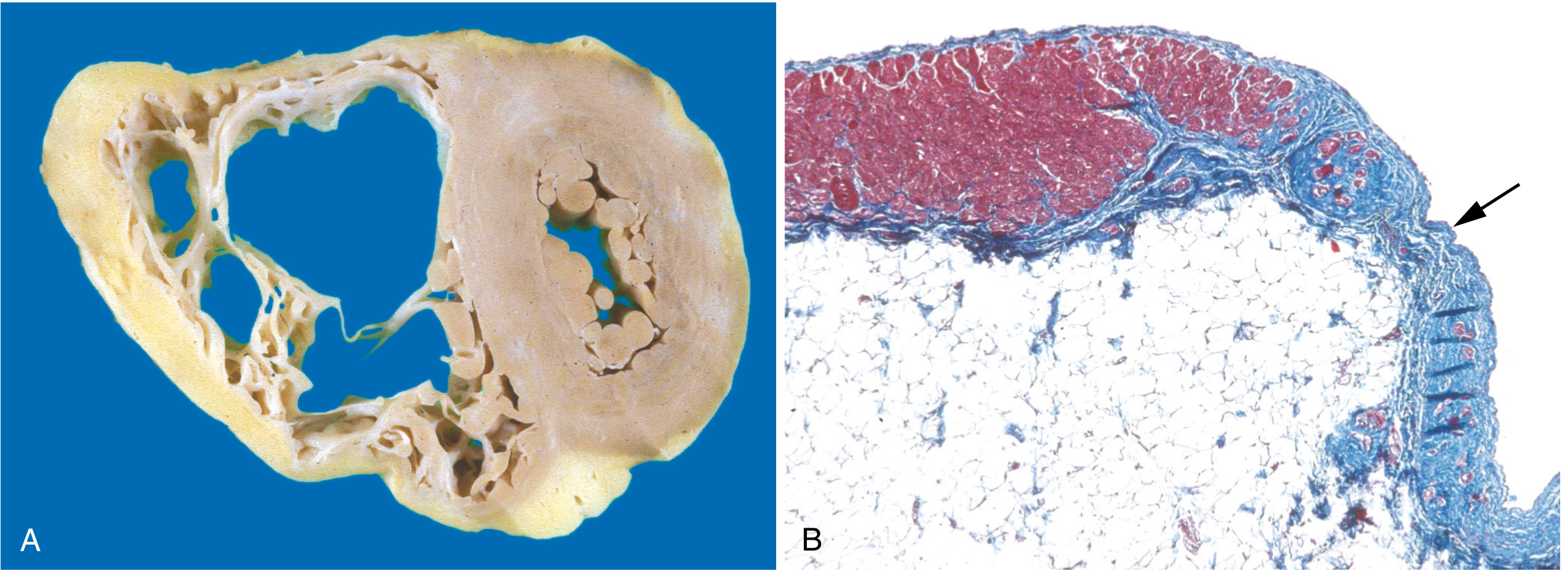
Genetics: Autosomal dominant, variable penetrance. Mutations predominantly in desmosomal junction proteins at the intercalated disk:
- Plakoglobin (JUP) - also causes Naxos syndrome (ARVC + palmoplantar keratoderma + woolly hair)
- Desmoplakin (DSP), Desmoglein-2 (DSG2), Desmocollin-2 (DSC2)
- PKP2 (plakophilin-2) is the most common mutation
ECG hallmarks: T-wave inversion in V1-V3; epsilon waves (delayed ventricular activation); VT with left bundle branch block morphology (right ventricular origin).
Diagnosis: Uses Task Force Criteria (major + minor) based on structural imaging (echo/MRI), tissue characterization, ECG repolarization/depolarization, arrhythmia pattern, and family history.
(Robbins & Cotran; Tintinalli's; Braunwald's Heart Disease)
5. Takotsubo (Stress) Cardiomyopathy
Also called "apical ballooning syndrome" or "broken heart syndrome."
- Trigger: Intense emotional or physical stress causes catecholamine surge
- Pattern: Transient apical and mid-ventricular ballooning with preserved or hyperkinetic basal function; mimics anterior STEMI
- Population: Predominantly postmenopausal women
- Course: Usually reversible within days to weeks
- Mechanism: Catecholamine-mediated myocardial stunning; coronary arteries are typically normal
(Braunwald's Heart Disease - chapter listing: Takotsubo Cardiomyopathy, p.1042)
6. Peripartum Cardiomyopathy
- Develops in the last month of pregnancy or within 5 months postpartum
- Presents as end-stage low-output heart failure
- No pre-existing cardiac disease
- Etiology: Likely multifactorial - anti-angiogenic factors (sFlt-1), prolactin cleavage products, myocarditis, immune mechanisms
- Prognosis variable; partial or full recovery possible in many patients
(Goldman-Cecil Medicine; Creasy & Resnik Maternal-Fetal Medicine)
7. Left Ventricular Non-Compaction (LVNC)
- Failure of the normal compaction of the ventricular myocardium during embryogenesis
- Prominent trabeculations with deep intertrabecular recesses in the LV
- Can overlap with DCM, HCM, or RCM phenotypes
- Risk of heart failure, arrhythmia, and thromboembolic events
- Genetic associations: TTN, MYH7, MYBPC3, and others
(Braunwald's Heart Disease)
Quick Comparison at a Glance
| Feature | DCM | HCM | RCM | ARVC |
|---|---|---|---|---|
| LV size | Dilated | Normal/small | Normal | Normal |
| Wall thickness | Thin/normal | Thick | Thick (infiltration) | RV thinned |
| Systolic function | Reduced | Normal/hyperdynamic | Normal (early) | RV reduced |
| Diastolic function | Impaired | Markedly impaired | Severely impaired | Impaired |
| Main risk | Heart failure, SCD | SCD (young athletes) | Heart failure | SCD, VT |
| Genetics | AD loss-of-function | AD gain-of-function | Variable | AD desmosomes |
Sources:
- Robbins, Cotran & Kumar Pathologic Basis of Disease (Cardiomyopathy, p.535-536)
- Braunwald's Heart Disease (Chapter 52 - Classification of Cardiomyopathies, p.1032-1049)
- Tintinalli's Emergency Medicine (Sudden Cardiac Death section, p.95)
- Goldman-Cecil Medicine (Cardiomyopathy chapter)
This is a shared conversation. Sign in to Orris to start your own chat.