Myocardial Infarction
Reading File
Asking for Preferences
Finding Sources
Searching PubMed
"myocardial infarction"[MeSH Terms] AND pathophysiology
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive content on definition, pathogenesis, types, and morphological progression of MI. Let me compile the full answer.
Myocardial Infarction - Overview
Definition
Myocardial infarction (MI), commonly called a "heart attack," is necrosis of cardiac muscle resulting from prolonged ischemia. The 2018 Universal Definition (joint task force of U.S. and European Cardiology groups) defines it as "the presence of acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia."
- ~800,000 MIs occur in the United States per year (nearly one every 40 seconds)
- ~400,000 deaths annually
- 10% occur before age 40; 45% before age 65
- Men are at higher risk than women, but the gap narrows with age; post-menopausal women lose estrogen-mediated protection, making IHD the #1 cause of death in older women
The major underlying cause is atherosclerosis of the coronary arteries.
Types of MI
By Depth of Necrosis
| Type | Description | Mechanism |
|---|---|---|
| Transmural | Necrosis spans the full wall thickness | Atherosclerosis + acute plaque rupture + thrombus (classic) |
| Subendocardial (Nontransmural) | Limited to inner 1/3 of the wall | Thrombus that lyses before full-thickness necrosis; or global hypoperfusion/shock |
| Multifocal Microinfarction | Scattered small foci | Small intramural vessel pathology (microemboli, vasculitis, cocaine-induced spasm) |
By Clinical/ECG Presentation
| Type | Pathophysiology | ECG Finding |
|---|---|---|
| STEMI (ST-elevation MI) | Complete thrombotic occlusion of epicardial vessel | ST segment elevation in affected leads |
| NSTEMI (Non-ST-elevation MI) | Partial/transient occlusion or subendocardial injury | ST depression or T-wave changes, no ST elevation |
Universal Classification (Types 1-5)
- Type 1 - Spontaneous MI from plaque rupture/erosion with thrombosis (classic atherothrombotic)
- Type 2 - Supply-demand mismatch (tachyarrhythmia, anemia, hypotension, vasospasm) without plaque rupture
- Type 3 - Sudden cardiac death before biomarkers can be drawn
- Type 4a/4b - MI associated with PCI or stent thrombosis
- Type 5 - MI associated with CABG
Pathophysiology
Step 1: Coronary Artery Occlusion
The sequence for a typical (Type 1) MI:
- An atheromatous plaque is eroded or suddenly disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents to circulating blood
- Platelets adhere, aggregate, and are activated - releasing thromboxane A2, ADP, and serotonin, causing further platelet aggregation and vasospasm
- Activation of coagulation via tissue factor exposure adds to the growing thrombus
- Within minutes, the thrombus can completely occlude the coronary artery lumen
Evidence: Angiography within 4 hours of MI onset shows coronary thrombosis in ~90% of cases. By 12-24 hours, this falls to ~60% - indicating that some occlusions clear spontaneously via thrombolysis or spasm relaxation.
In ~10% of cases, MI occurs without typical atherothrombosis, via:
- Vasospasm (with/without atherosclerosis) - cocaine, ephedrine
- Embolism (from atrial fibrillation mural thrombus, endocarditis vegetations, prosthetic material, or patent foramen ovale)
- Small vessel disorders: vasculitis, sickle cell disease, amyloid deposition, vascular dissection
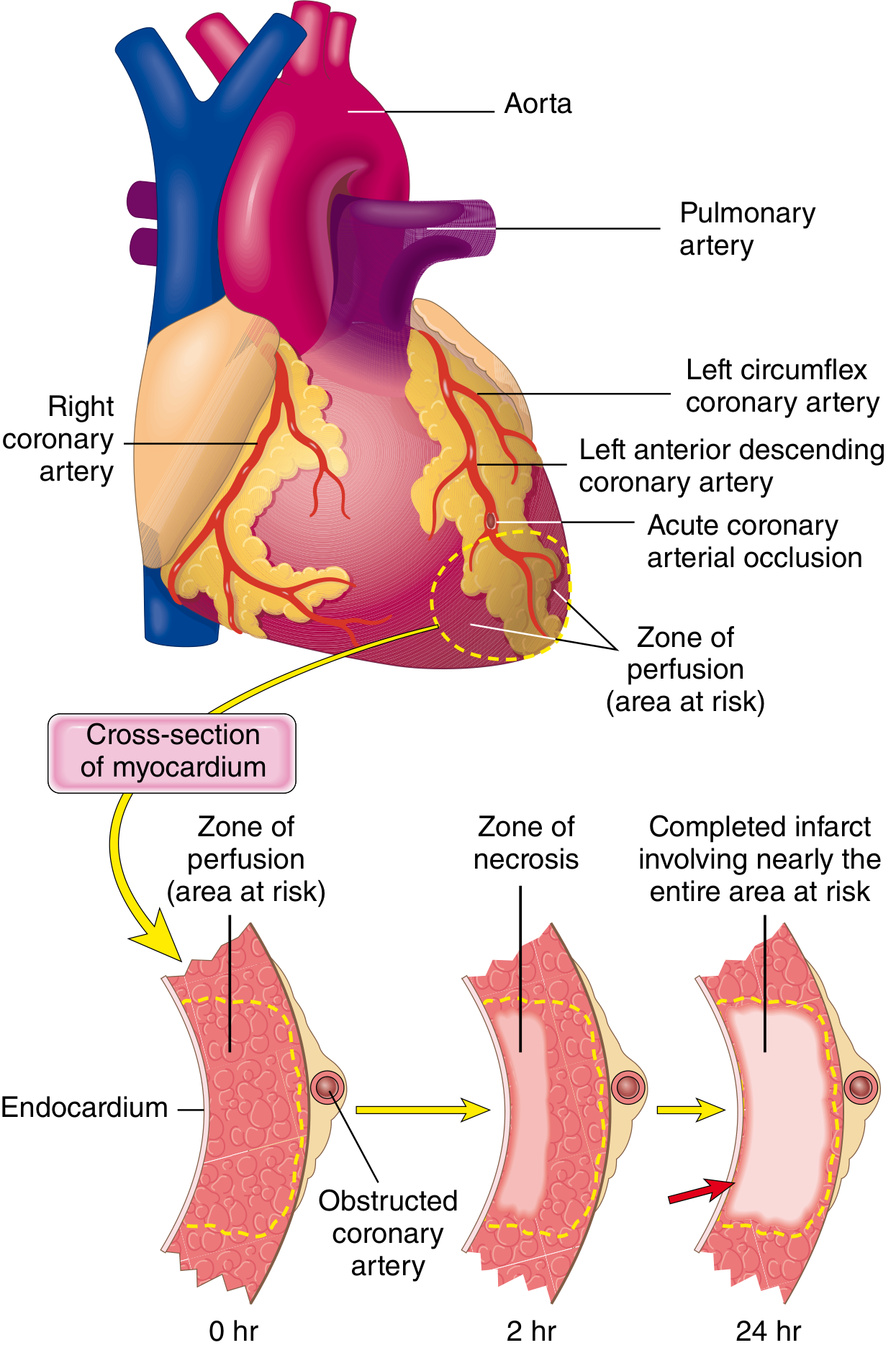
Step 2: Myocardial Response to Ischemia
Loss of blood supply triggers a cascade of functional, biochemical, and morphological consequences:
| Event | Time |
|---|---|
| Onset of ATP depletion | Seconds |
| Loss of contractility | < 2 minutes |
| ATP reduced to 50% of normal | 10 minutes |
| ATP reduced to 10% of normal | 40 minutes |
| Irreversible cell injury (necrosis) | 20-40 minutes |
| Microvascular injury | > 1 hour |
Key mechanisms:
- Cessation of aerobic metabolism → inadequate ATP production → accumulation of lactic acid
- Loss of contractility within minutes (before cell death - this is reversible)
- Sarcolemmal membrane disruption allows intracellular proteins (troponin, CK-MB) to leak into circulation - basis of biomarker testing
- Only ischemia of 20-40 minutes or longer (with blood flow ≤10% of normal) leads to irreversible necrosis
The "Wavefront Phenomenon"
Necrosis begins in the subendocardial zone first - the region most vulnerable because:
- It is the last area to receive blood from epicardial vessels
- Exposed to the highest intramural pressures, impeding blood inflow
With prolonged ischemia, a wavefront of cell death progresses centripetally (outward toward the epicardium), driven by edema, reactive oxygen species, and inflammatory mediators. A narrow rim (~0.1 mm) of subendocardial myocardium immediately under the endocardium is spared by direct diffusion from the ventricular lumen.
Therapeutic implication: The time-dependent wavefront is why "time is muscle" - early reperfusion (thrombolysis or angioplasty) limits the extent of the infarct.
Step 3: Vascular Territory and Location
In a typical right-dominant circulation (~80% of people):
| Artery Occluded | Frequency | Zone of Infarction |
|---|---|---|
| LAD (Left Anterior Descending) | 40-50% | Anterior LV wall, anterior 2/3 of septum, apex |
| RCA (Right Coronary Artery) | 30-40% | Inferior/posterior LV wall, posterior 1/3 of septum, RV free wall |
| LCX (Left Circumflex) | 15-20% | Lateral wall of LV |
Morphological Progression (Gross & Microscopic)
| Time | Gross Appearance | Microscopic Changes |
|---|---|---|
| 0-0.5 hr | None visible | None visible (wavy fiber change may begin) |
| 1-4 hr | None (TTC stain shows pale zone) | Early coagulation necrosis; edema; hemorrhage |
| 4-12 hr | Dark mottling | Coagulation necrosis; beginning neutrophil infiltration |
| 12-24 hr | Dark mottling | Ongoing necrosis; pyknosis of nuclei; prominent neutrophils |
| 1-3 days | Mottling with yellow-tan center | Necrosis with loss of nuclei; dense neutrophil infiltrate |
| 3-7 days | Hyperemic border; softening center | Macrophage infiltration begins; phagocytosis of dead cells |
| 1-3 weeks | Yellow-tan, soft; vascularized edges | Granulation tissue; progressive fibrosis |
| Weeks-months | White-gray scar (contracted) | Dense collagen scar; minimal cellularity |
MIs < 12 hours old are often not visible on gross examination. The triphenyl tetrazolium chloride (TTC) stain reveals early infarcts: viable myocardium stains brick-red; infarcted (enzyme-depleted) tissue appears as an unstained pale zone.
Reperfusion Injury
When perfusion is restored before irreversible injury occurs, myocardium can be preserved. However:
- Stunned myocardium - reperfused myocardium may be profoundly non-contractile for days despite viability (persistent biochemical abnormalities)
- Reperfusion injury - reactive oxygen species and calcium overload cause additional damage at the moment of reperfusion
- Arrhythmias - ischemic regions show electrical instability; in 80-90% of cases, cardiac death in MI is due to ventricular fibrillation (not mechanical failure)
Summary
Myocardial infarction is fundamentally a consequence of coronary atherosclerosis complicated by plaque rupture and thrombosis, leading to prolonged ischemia and irreversible cardiomyocyte necrosis. The three major patterns are transmural (STEMI), subendocardial (NSTEMI), and microinfarction. The time-critical nature of myocardial salvage is driven by the 20-40 minute threshold for irreversible injury and the centripetal wavefront of necrosis.
Sources: Robbins & Cotran Pathologic Basis of Disease (9780443264528); Robbins & Kumar Basic Pathology (9780323790185); Ganong's Review of Medical Physiology (9781260122404)
Myocardial Infarction
Asking for Preferences
The previous session covered this topic in full. Here is the complete overview directly from that research:
Myocardial Infarction - Overview, Types & Pathophysiology
Definition
Myocardial infarction (MI), commonly called a "heart attack," is necrosis of cardiac muscle resulting from prolonged ischemia. The 2018 Universal Definition (joint U.S./European Cardiology task force) defines it as "the presence of acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia."
Epidemiology:
- ~800,000 MIs per year in the United States (one every ~40 seconds)
- ~400,000 deaths annually
- 10% occur before age 40; 45% before age 65
- Men at higher risk than women until menopause, after which the gap closes - IHD is the #1 cause of death in older women
Types of MI
By Depth of Necrosis
| Type | Description | Mechanism |
|---|---|---|
| Transmural | Full wall thickness necrosis | Atherosclerosis + acute plaque rupture + occlusive thrombus |
| Subendocardial (Nontransmural) | Inner 1/3 of wall only | Thrombus that lyses before full-thickness necrosis; or global hypoperfusion/shock |
| Multifocal Microinfarction | Scattered small foci | Small intramural vessel disease (microemboli, vasculitis, cocaine spasm) |
By ECG / Clinical Presentation
| Type | Pathophysiology | ECG |
|---|---|---|
| STEMI | Complete occlusion of epicardial vessel | ST elevation in affected leads |
| NSTEMI | Partial/transient occlusion, subendocardial injury | ST depression, T-wave changes; no ST elevation |
Universal Classification (Types 1-5)
| Type | Mechanism |
|---|---|
| Type 1 | Spontaneous - plaque rupture/erosion with thrombosis (classic atherothrombotic) |
| Type 2 | Supply-demand mismatch (tachyarrhythmia, anemia, hypotension, vasospasm) without plaque rupture |
| Type 3 | Sudden cardiac death before biomarkers obtainable |
| Type 4a/4b | MI associated with PCI or stent thrombosis |
| Type 5 | MI associated with CABG |
Pathophysiology
1. Coronary Artery Occlusion (Typical Type 1 Sequence)
- An atheromatous plaque is eroded or suddenly disrupted by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents to blood
- Platelets adhere, aggregate, and activate - releasing thromboxane A₂, ADP, and serotonin, driving further aggregation and vasospasm
- Coagulation cascade activation via tissue factor exposure amplifies the growing thrombus
- Within minutes, the thrombus can completely occlude the coronary lumen
Angiography within 4 hours of MI onset shows thrombotic occlusion in ~90% of cases. By 12-24 hours this falls to ~60%, as some occlusions clear spontaneously through lysis or spasm relaxation - the basis for thrombolytic therapy.
Non-atherothrombotic causes (~10% of MIs):
- Vasospasm (cocaine, ephedrine, or idiopathic)
- Embolism (from atrial fibrillation mural thrombus, endocarditis vegetations, prosthetic material, patent foramen ovale)
- Small vessel disease: vasculitis, sickle cell disease, amyloid deposition, vascular dissection
- Severe fixed atherosclerosis + increased demand (tachycardia, hypertension) - causes subendocardial infarction without frank thrombosis
2. Cellular Response to Ischemia
| Event | Time |
|---|---|
| Onset of ATP depletion | Seconds |
| Loss of contractility | < 2 minutes |
| ATP reduced to 50% of normal | 10 minutes |
| ATP reduced to 10% of normal | 40 minutes |
| Irreversible necrosis (point of no return) | 20-40 minutes |
| Microvascular injury | > 1 hour |
Biochemical cascade:
- Cessation of aerobic metabolism → ATP fails → lactic acid accumulates
- Loss of myocardial contractility within 1-2 minutes (reversible at this stage)
- Sarcolemmal membrane disruption → intracellular proteins (troponin, CK-MB) leak into interstitium and circulation - the basis of biomarker testing
- Only ischemia lasting ≥20-40 minutes with blood flow ≤10% of normal produces irreversible coagulative necrosis
3. The "Wavefront Phenomenon" - How Necrosis Spreads
Necrosis begins in the subendocardial zone and spreads outward (centripetally toward the epicardium):
Why the subendocardium is most vulnerable:
- Last region to receive blood delivered via epicardial vessels
- Exposed to the highest intramural pressures, impeding blood inflow
A narrow rim (~0.1 mm) immediately under the endocardium is spared by direct oxygen diffusion from the ventricular lumen.
Clinical implication: This wavefront is why "time is muscle" - early reperfusion (PCI or thrombolysis) interrupts the wavefront and limits infarct size.
4. Coronary Territory and Infarct Location
In the typical right-dominant circulation (~80% of individuals):
| Artery | Frequency | Territory Infarcted |
|---|---|---|
| LAD (Left Anterior Descending) | 40-50% | Anterior LV wall, anterior 2/3 of septum, apex |
| RCA (Right Coronary Artery) | 30-40% | Inferior/posterior LV wall, posterior 1/3 of septum, posterior RV free wall |
| LCX (Left Circumflex) | 15-20% | Lateral wall of LV |
- Left main coronary artery occlusion is rare but catastrophic
- 15-30% of RCA occlusions extend into the right ventricular wall
- Isolated RV infarction is unusual (1-3% of cases)
Morphological Progression
| Time | Gross Appearance | Microscopic Changes |
|---|---|---|
| 0-0.5 hr | None visible | None visible (wavy fiber change may begin) |
| 1-4 hr | None (TTC stain shows pale zone) | Early coagulation necrosis; edema |
| 4-12 hr | Dark mottling | Coagulation necrosis; early neutrophil infiltration |
| 12-24 hr | Dark mottling | Nuclear pyknosis; dense neutrophil infiltrate |
| 1-3 days | Yellow-tan center, hyperemic border | Loss of nuclei; macrophage infiltration begins |
| 3-7 days | Softening, yellow center, red border | Macrophage phagocytosis; granulation tissue begins |
| 1-3 weeks | Yellow-tan, soft; vascularized edges | Active granulation tissue; progressive fibrosis |
| Weeks-months | White-gray contracted scar | Dense collagen scar; minimal cellularity |
MIs < 12 hours old are often invisible on gross examination. The triphenyl tetrazolium chloride (TTC) stain is used to detect early infarcts: viable myocardium stains brick-red (preserved dehydrogenase activity); infarcted tissue appears as an unstained pale zone (enzymes have leaked out).
Reperfusion and Its Consequences
Restoration of blood flow before irreversible injury preserves myocardium - the goal of all acute MI therapy. However, reperfusion has its own consequences:
- Stunned myocardium - viable but non-contractile for days after reperfusion, due to persistent biochemical dysfunction; may produce transient but reversible cardiac failure
- Reperfusion injury - reactive oxygen species and calcium overload at the moment of reperfusion cause additional myocyte death
- Arrhythmias - ischemic regions develop electrical instability; in 80-90% of cases, cardiac death in MI is from ventricular fibrillation, not mechanical failure
- No-reflow phenomenon - microvascular injury after >1 hour of ischemia can prevent tissue reperfusion even after the epicardial vessel is opened
Key Takeaways
- MI = irreversible ischemic necrosis of cardiomyocytes, threshold at 20-40 minutes of severe ischemia
- The dominant mechanism is plaque rupture → platelet aggregation → thrombus → occlusion
- Necrosis spreads as a subendocardial-to-epicardial wavefront over 6-12 hours
- Three anatomical patterns: transmural (STEMI), subendocardial (NSTEMI), microinfarction
- Five universal types by mechanism (Type 1-5), with Type 1 atherothrombotic being the most common
- Biomarker release (troponin, CK-MB) reflects sarcolemmal disruption and is the clinical cornerstone of diagnosis
Sources: Robbins & Cotran Pathologic Basis of Disease; Robbins & Kumar Basic Pathology; Ganong's Review of Medical Physiology - via Medical Textbook Library
This is a shared conversation. Sign in to Orris to start your own chat.