methemoglobinemias
Reading File
Finding Sources
Searching PubMed
methemoglobinemia
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
I now have comprehensive information from multiple authoritative textbooks plus great images. Let me compile the full answer.
Methemoglobinemias
Definition
Methemoglobinemia is a condition in which hemoglobin iron is oxidized from the normal ferrous (Fe²+) state to the ferric (Fe³+) state. Ferric hemoglobin (methemoglobin) cannot bind oxygen for transport. Normally, <1-2% of circulating hemoglobin exists as methemoglobin; higher concentrations define the clinical condition.
- Goldman-Cecil Medicine, 26e
Pathophysiology
Normal red cells continuously generate small amounts of methemoglobin via oxidant stress. Two enzymatic systems reduce it back to functional hemoglobin:
Primary pathway (responsible for ~95% of reduction):
- Glycolysis produces NADH
- NADH donates electrons to cytochrome b5, which then reduces Fe³+ back to Fe²+
- Enzyme: cytochrome b5 reductase (also called NADH-methemoglobin reductase or diaphorase I)
Secondary pathway (~5% under normal conditions, but critical for antidote action):
- Hexose monophosphate shunt generates NADPH
- NADPH-methemoglobin reductase then reduces methemoglobin
- This pathway is normally minor, but methylene blue activates it therapeutically
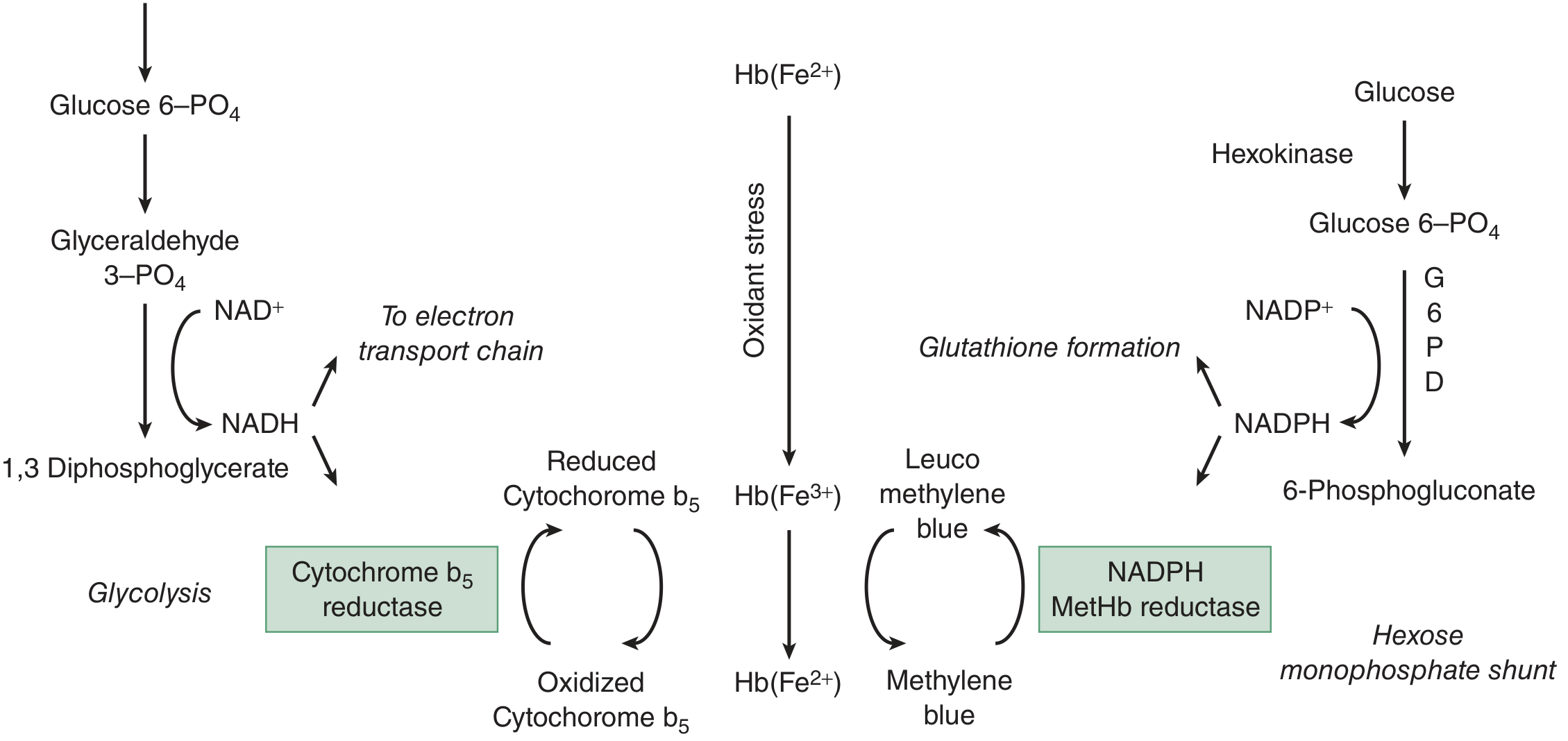
Note on G6PD: Because NADPH comes from the hexose monophosphate shunt via G6PD, patients with G6PD deficiency have impaired NADPH production - this does NOT increase their risk of methemoglobinemia (the primary NADH pathway is intact), but it does:
- Make methylene blue ineffective (cannot generate leuco-methylene blue without NADPH)
- Increase risk of hemolysis on oxidant exposure
Non-enzymatic reducers (vitamin C, glutathione) play only a minor role.
Why methemoglobin is worse than equivalent anemia:
With 50% methemoglobin, the oxyhemoglobin dissociation curve shifts leftward - the remaining hemoglobin holds on tighter to oxygen, worsening tissue oxygen delivery beyond the simple reduction in oxygen-carrying capacity.
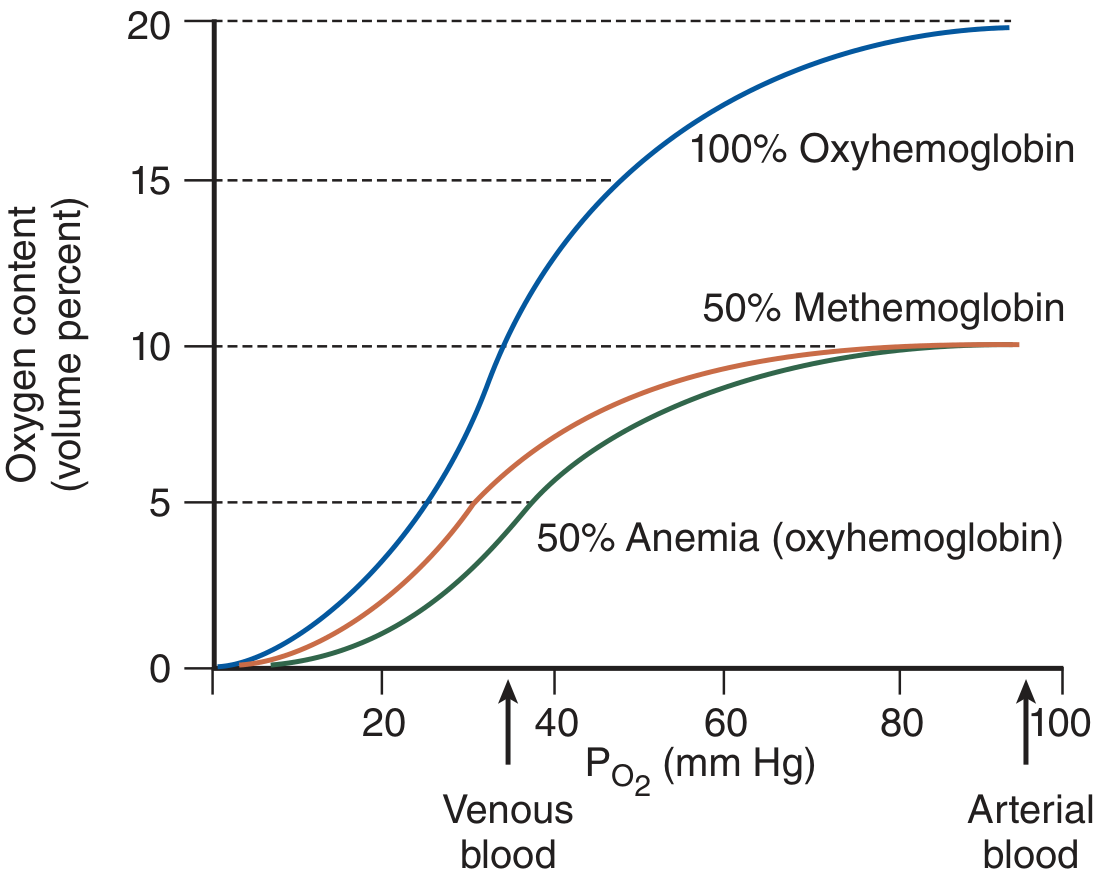
- Tintinalli's Emergency Medicine, p. 1371-1372
Classification & Causes
1. Acquired (Most Common)
Occurs when exogenous oxidants overwhelm cytochrome b5 reductase:
| Category | Agents | Notes |
|---|---|---|
| Local anesthetics | Benzocaine | Most commonly reported; occurs with topical sprays (e.g., Cetacaine for endoscopy) |
| Prilocaine | Common in topical anesthetics; EMLA generally safe at recommended doses | |
| Lidocaine, Dibucaine | Rare | |
| Antimicrobials | Dapsone | Hydroxylamine metabolite; can be inhibited by cimetidine |
| Sulfamethoxazole | Uncommon | |
| Antimalarials (primaquine) | Common | |
| Nitrofurantoin | ||
| Analgesics | Phenazopyridine (Pyridium) | Commonly reported |
| Phenacetin | Rarely used now | |
| Nitrates/Nitrites | Amyl nitrite, isobutyl nitrite | Cyanide antidote / recreational use |
| Sodium nitrite | Cyanide antidote kit (therapeutic goal) | |
| Well water (nitrate runoff) | Major problem in infants | |
| Nitroglycerin | Rare | |
| Other | Rasburicase | Hyperuricemia / tumor lysis syndrome |
| Silver nitrate | Excessive topical use |
Special susceptibility in neonates/infants: Underdeveloped cytochrome b5 reductase makes infants highly susceptible - from nitrogenous vegetables (spinach), high-nitrate well water, or GI flora converting nitrates to nitrites in gastroenteritis.
2. Congenital
A. Cytochrome b5 reductase deficiency (autosomal recessive)
- Type I: Enzyme deficiency limited to red cells only. Methemoglobin 20-40%. Cyanosis present from birth, but patients are otherwise asymptomatic and develop normally.
- Type II: Generalized enzyme deficiency (affects all tissues including CNS). Associated with persistent neurologic features - severe intellectual disability, microcephaly. Much rarer and more serious.
B. Hemoglobin M variants
- Point mutations in α- or β-globin chains stabilize iron in the ferric state
- Only viable in the heterozygous form (homozygous is lethal)
- Patients develop profound cyanosis but tolerate elevated methemoglobin due to compensatory mechanisms
- Do NOT respond to methylene blue
Clinical Features by Methemoglobin Level
| MetHb Level | Symptoms |
|---|---|
| <10% | Usually asymptomatic; cyanosis may begin to appear |
| 10-20% | Cyanosis (chocolate-brown blood, gray-blue skin). Often few symptoms. |
| 20-30% | Anxiety, headache, weakness, lightheadedness, tachypnea, sinus tachycardia |
| 30-50% | Fatigue, confusion, worsening symptoms |
| 50-60% | Myocardial ischemia, dysrhythmias, depressed mental status, coma, seizures, lactic acidosis |
| >70% | Largely incompatible with life |
Key clinical pearl: Congenital methemoglobinemia patients tolerate levels of 30-40% well due to adaptation - their primary problem is cosmetic/social cyanosis. Acute toxic methemoglobinemia at the same levels can be life-threatening.
Cyanosis threshold: Detectable at a methemoglobin concentration of 1.5 g/dL (corresponding to ~10-15% in a non-anemic person). Compare: deoxyhemoglobin requires ~5 g/dL to cause cyanosis. This means methemoglobinemia causes visible cyanosis at a level when oxygen-carrying capacity is still ~90% of normal - so cyanotic patients may look worse than they feel.
The classic presentation: Cyanosis that does not improve with supplemental oxygen + chocolate-brown arterial blood.
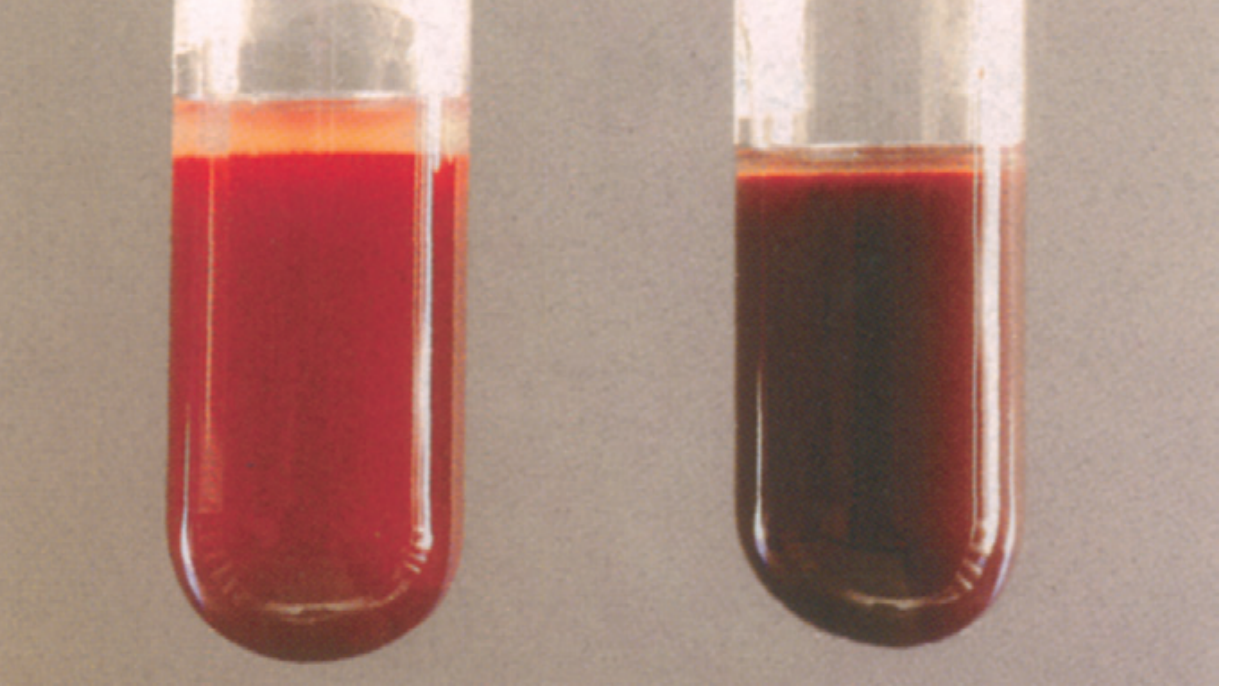
- Pfenninger & Fowler's Procedures for Primary Care, p. 55
Diagnosis
- Clinical suspicion - cyanosis unresponsive to O2, history of oxidant exposure
- Arterial blood gas - SpO2 will be normal or near-normal (pulse oximetry reads MetHb as ~85% saturation - falsely reassuring or falsely alarming depending on actual SpO2). ABG shows normal PaO2 but co-oximetry reveals elevated MetHb %.
- Co-oximetry (multi-wavelength oximetry) - directly quantifies MetHb; most hospital blood gas machines can do this
- Blood color - chocolate-brown blood that does NOT turn red on air/O2 exposure (distinguishes from deoxyhemoglobin, which does turn red)
- KCN test - Adding potassium cyanide to the blood sample turns it red in methemoglobinemia but NOT in hemoglobin M (useful to distinguish them)
Treatment
Acute/Acquired Methemoglobinemia
1. Remove the offending agent
2. Supplemental oxygen (high-flow, though it won't resolve methemoglobin - gives more substrate for remaining functional Hb)
3. Methylene blue (first-line antidote)
- Dose: 1-2 mg/kg IV over 5 minutes; may repeat in 30-60 minutes if needed
- Higher doses (up to 6-7 mg/kg) may be required in severe cases
- Mechanism: Acts as a cofactor for NADPH-methemoglobin reductase. Methylene blue is reduced to leuco-methylene blue (using NADPH), which then reduces MetHb back to Hb.
- Contraindications/cautions:
- G6PD deficiency - methylene blue will be ineffective (no NADPH) and may actually worsen hemolysis. Consider ascorbic acid or exchange transfusion instead.
- Renal insufficiency (increased methylene blue concentrations)
- Paradox at high doses: Methylene blue itself can cause methemoglobinemia at doses >7 mg/kg
- Drug interactions: Serotonin syndrome risk with SSRIs, SNRIs, clomipramine, bupropion (methylene blue is an MAO inhibitor - this interaction is considered contraindicated)
- Side effects: nausea, vomiting, headache, blue-green urine/feces, stained skin
- Dilute ProvayBlue in D5W (not saline - reduces solubility)
4. For dapsone: Cimetidine inhibits the formation of dapsone's toxic hydroxylamine metabolite and can reduce ongoing methemoglobin generation as adjunctive therapy.
Congenital Methemoglobinemia
- Type I cytochrome b5 reductase deficiency (cosmetic cyanosis): Ascorbic acid (vitamin C) - reduces methemoglobin via non-enzymatic pathway; also methylene blue for more rapid correction
- Hemoglobin M: No treatment needed (well tolerated); methylene blue is ineffective
Exchange transfusion
-
Reserve for patients with G6PD deficiency or when methylene blue is ineffective/contraindicated
-
Tintinalli's Emergency Medicine, p. 1371-1373; Goldman-Cecil Medicine; Harriet Lane Handbook (methylene blue dosing)
Summary Table: Key Differentiators
| Feature | Methemoglobinemia | Oxyhemoglobin desaturation (true hypoxia) |
|---|---|---|
| Cyanosis with O2 | Persists | Improves |
| Blood color | Chocolate-brown | Dark red/purple |
| PaO2 on ABG | Normal | Low |
| Pulse oximetry | ~85% (spuriously) | Accurately low |
| Response to methylene blue | Yes (if not G6PD def.) | No |
Recent literature note: A 2024 systematic review (PMID 37468669) specifically examined rasburicase-induced methemoglobinemia/hemolytic anemia, confirming this as a clinically important drug-induced cause - relevant to oncology patients receiving treatment for tumor lysis syndrome. A 2024 meta-analysis (PMID 39331646) used methemoglobin as a surrogate marker for primaquine antihypnozoite activity in vivax malaria, underscoring its clinical relevance in antimalarial therapy monitoring.
fibroblasts
Reading File
Finding Sources
Searching PubMed
fibroblast biology function connective tissue
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
I have comprehensive content from multiple authoritative textbooks. Now I'll compile the full answer.
Fibroblasts
Overview
Fibroblasts are the most common cells in connective tissue proper and the principal architects of the extracellular matrix (ECM). Their name reflects their dual nature: fibro- (fiber-producing) and -blast (immature/active cell). They are found in virtually every organ and tissue, where they maintain structural integrity, respond to injury, and regulate tissue remodeling.
- Junqueira's Basic Histology, 17e
Origin & Lineage
Fibroblasts differentiate from resting mesenchymal cells (of mesodermal origin) in connective tissue. They do not arrive at wound sites by diapedesis from circulating blood - they are tissue-resident cells that are activated locally. Bone marrow-derived mesenchymal stem cells (MSCs) can also differentiate into fibroblasts, particularly at injury sites, though this contribution appears to be a minor percentage in most small wounds.
Morphology: Active vs. Quiescent
Two states can be distinguished histologically (H&E stain):
Active fibroblast (fibroblast proper):
- Larger cell with abundant, irregularly branched cytoplasm
- Large, ovoid, euchromatic nucleus with a prominent nucleolus
- Rich in rough endoplasmic reticulum (RER) and a well-developed Golgi apparatus
- Basophilic cytoplasm (reflecting heavy protein synthesis)
Quiescent fibroblast (fibrocyte):
- Smaller with fewer cytoplasmic extensions
- Smaller, darker, heterochromatic nucleus
- Much less RER
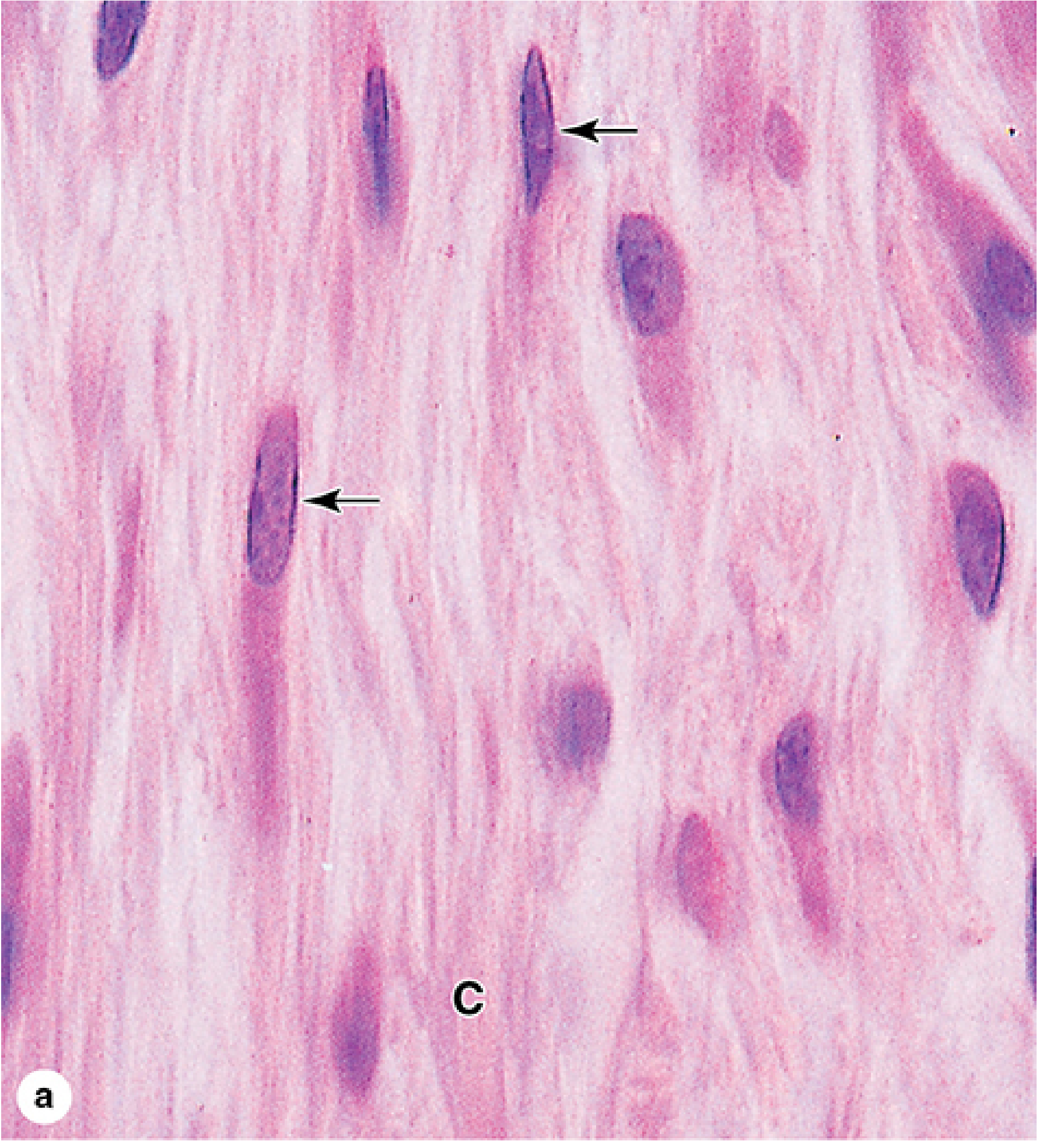
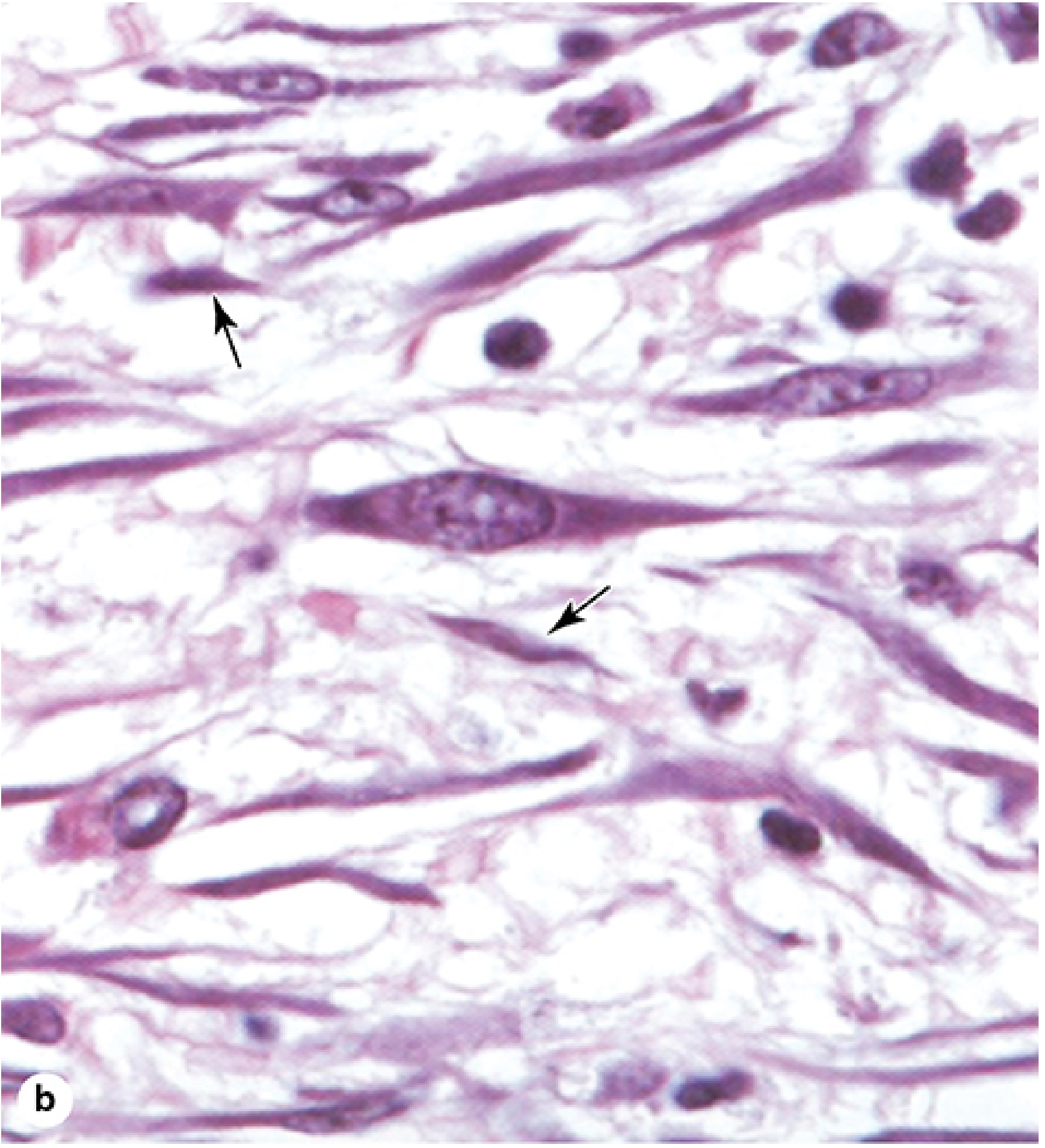
- Junqueira's Basic Histology, 17e, p. 261-263
ECM Products Synthesized by Fibroblasts
Fibroblasts are the main source of nearly every ECM component:
| Product | Notes |
|---|---|
| Collagens (types I, III, V, VI) | Most abundant protein in the body; primary structural scaffold |
| Elastin | Provides recoil/elasticity to tissues |
| Glycosaminoglycans (GAGs) | e.g., hyaluronic acid, chondroitin sulfate |
| Proteoglycans | GAGs covalently linked to core proteins (decorin, versican, aggrecan) |
| Multiadhesive glycoproteins | Fibronectin, laminin - mediate cell-ECM adhesion |
| Matrix metalloproteinases (MMPs) | Collagenase (MMP-1) for ECM degradation and remodeling |
| Growth factors/cytokines | FGF-2 (autocrine), TGF-β, IL-6, etc. |
Most secreted ECM components undergo further post-translational modification outside the cell (e.g., procollagen cleavage, collagen cross-linking via lysyl oxidase) before final matrix assembly.
Growth Factor Regulation
In adult tissues, fibroblasts are normally arrested in G0 phase and rarely divide. They respond to growth factors that drive re-entry into the cell cycle:
- PDGF - major mitogen; released by platelets and macrophages at injury sites
- TGF-β - stimulates fibroblast proliferation indirectly (via PDGF release) and drives ECM synthesis
- FGF-2 - autocrine stimulation of fibroblast replication
- EGF / IGF-1 - required for continued proliferation
- TNF-α, C5 fragments, thrombin, leukotriene B4 - chemotactic signals drawing fibroblasts to wound sites
Importantly, fibroblasts do not require growth factors to survive - they can live quiescently in growth factor-free conditions.
- Sabiston Textbook of Surgery, p. 394-395
Fibroblasts in Wound Healing
Fibroblast involvement in healing proceeds in defined phases:
1. Lag phase (days 0-3/5): Undifferentiated mesenchymal cells differentiate into specialized fibroblasts in response to PDGF, TGF-β, and other signals from platelets and macrophages. No significant collagen deposition occurs during this window.
2. Proliferative/fibroplasia phase: Fibroblasts proliferate, migrate into the wound using fibronectin and fibrin as scaffolding, and begin producing collagen (initially type III, later type I).
3. Maturation/remodeling phase: Collagen synthesis peaks then declines, eventually balancing collagenase (MMP-1) activity. Glycoprotein and mucopolysaccharide levels decrease. New capillaries regress. This phase continues for months to years.
Myofibroblast Differentiation
A key specialized state: when fibroblasts differentiate into myofibroblasts, they acquire smooth muscle cell-like features:
- De novo expression of α-smooth muscle actin (α-SMA) - the defining marker
- Contractile stress fibers enabling wound contraction
- Driven by TGF-β1 and the fibronectin splice variant ED-A
- Rho-ROCK signaling integrates mechanical tension and integrin/FAK signaling
After wound closure, myofibroblasts normally undergo apoptosis or reverse differentiation back to fibroblasts. Abnormal persistence of myofibroblasts leads to:
-
Hypertrophic scars
-
Keloids
-
Fibrotic diseases (pulmonary fibrosis, hepatic cirrhosis, systemic sclerosis)
-
Dermatology 5e, p. [block30]; Sabiston Surgery
Fibroblast Subsets (Single-Cell Era)
Single-cell transcriptomic studies have revealed that fibroblasts are not a homogeneous population. In the synovium (well-studied in rheumatoid arthritis), at least four discrete fibroblast clusters have been identified:
| Cluster | Markers | Function |
|---|---|---|
| Lining layer | CD55+ (SC-F4) | Line the synovial joint surface |
| Sublining CD34+ | CD34+ (SC-F1) | Perivascular; regulatory |
| Sublining HLA-DRAhi | HLA-DRAhi (SC-F2) | Antigen presentation-related |
| Sublining DKK3+ | DKK3+ (SC-F3) | Matrix production |
The positional identity of these subsets is determined by NOTCH signaling gradients from adjacent endothelial cells. In RA, the fibroblast activation protein (FAP) marks pathologic clusters, with THY1 further subdividing them into:
- THY1+ FAP+: drives leukocyte recruitment
- THY1- FAP+: drives joint destruction and cartilage erosion
These insights are now being exploited for selective therapeutic targeting.
- Firestein & Kelley's Textbook of Rheumatology, p. 312
Fibroblasts in Specific Tissues
Synovial fibroblasts (Rheumatology)
In RA, synovial fibroblasts (FLS - fibroblast-like synoviocytes) become pathologically activated, exhibiting invasive, tumor-like behavior: they proliferate within the pannus, invade cartilage and bone, and resist apoptosis. They are a primary therapeutic target.
Adventitial fibroblasts (Cardiology)
In the vessel wall adventitia, fibroblasts produce collagen, elastin, and inflammatory mediators (IL-6, MCP-1, NF-κB). Warburg metabolism (aerobic glycolysis) drives their proliferation, migration, and inflammatory cell influx - contributing to fibrosis and vascular stiffening in cardiovascular disease.
- Braunwald's Heart Disease, 11e
Pulmonary fibroblasts
Activated pulmonary fibroblasts and myofibroblasts are central to the pathogenesis of idiopathic pulmonary fibrosis (IPF), driven by TGF-β1 signaling.
Dermal fibroblasts
Establish resting ECM tension during development. During repair, they reconstitute the dermis. Fibroblasts from different anatomical sites (e.g., palatal vs. skin) differ in their intrinsic gene expression, explaining differences in scarless healing at certain sites.
Disease Associations
| Disease | Fibroblast Role |
|---|---|
| Rheumatoid arthritis | Invasive, cartilage-eroding synovial fibroblasts (FLS) |
| Systemic sclerosis (scleroderma) | Constitutively activated myofibroblasts; excessive collagen |
| Idiopathic pulmonary fibrosis | TGF-β-driven myofibroblast accumulation |
| Keloids / hypertrophic scars | Persistent myofibroblasts post-wound healing |
| Cancer | Cancer-associated fibroblasts (CAFs) remodel tumor stroma, promote invasion and immunosuppression |
| Liver cirrhosis | Hepatic stellate cells (hepatic fibroblast-like cells) as myofibroblasts |
| Dupuytren's contracture | Nodular myofibroblasts in palmar fascia |
Key Surface Markers
| Marker | Notes |
|---|---|
| Vimentin | Pan-fibroblast intermediate filament; most reliable general marker |
| α-SMA (ACTA2) | Marks activated myofibroblasts |
| FAP (fibroblast activation protein) | Cancer-associated fibroblasts and RA-FLS |
| FSP-1 (S100A4) | Fibroblast-specific protein-1 |
| CD90 (THY1) | Sublining synovial fibroblasts; also distinguishes subsets |
| CD34 | Perivascular/sublining fibroblast subset |
| Cadherin-11 | Synovial fibroblasts; mediates homotypic aggregation |
Recent research note: A 2023 review (PMID 37646029) highlighted fibroblasts as "cellular choreographers" of wound healing, emphasizing their dynamic cross-talk with immune cells - macrophages, mast cells, and T cells - as an emerging therapeutic frontier in regenerative medicine.
types of apolipoproteins
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
I have rich, comprehensive data from multiple authoritative sources. Now composing the full answer.
Apolipoproteins
Apolipoproteins (apos) are the protein components of lipoprotein particles. They serve three broad physiologic functions:
- Activating or inhibiting key enzymes in lipid metabolism
- Maintaining structural integrity of the lipoprotein particle (via amphipathic helices that interface between the hydrophobic lipid core and the aqueous plasma)
- Facilitating cellular uptake of lipoproteins by acting as ligands for specific cell-surface receptors
Most apolipoproteins (except apoB and apo(a)) can freely exchange between different lipoprotein particles during circulation. ApoB and apo(a) are permanently embedded due to their beta-sheet structures which have much higher lipid affinity than amphipathic helices alone.
- Tietz Textbook of Laboratory Medicine, 7th ed.
Master Reference Table
| Apolipoprotein | MW (Da) | Chromosome | Lipoprotein Carrier(s) | Primary Function |
|---|---|---|---|---|
| ApoA-I | 29,016 | 11 | Chylomicron, HDL | Cofactor of LCAT; ABCA1 ligand (cholesterol efflux) |
| ApoA-II | 17,414 | 1 | HDL | Inhibits hepatic lipase; unclear role |
| ApoA-IV | 44,465 | 11 | Chylomicron, HDL | Activates LCAT; facilitates LPL action; satiety signaling |
| ApoA-V | ~39,000 | 11 | VLDL, HDL | Reduces TG by enhancing LPL function; modulates VLDL secretion |
| ApoB-100 | 512,723 | 2 | VLDL, IDL, LDL, Lp(a) | Secretion of TG from liver; ligand for LDL receptor (LDLR) |
| ApoB-48 | 240,800 | 2 | Chylomicron | Secretion of TG from intestine |
| ApoC-I | 6,630 | 19 | Chylomicron, VLDL, HDL | Activates LCAT; inhibits chylomicron clearance |
| ApoC-II | 8,900 | 19 | Chylomicron, VLDL, HDL | Essential cofactor of lipoprotein lipase (LPL) |
| ApoC-III | 8,800 | 11 | Chylomicron, VLDL, LDL, HDL | Inhibits LPL; inhibits clearance of TG-rich lipoprotein remnants |
| ApoE | 34,145 | 19 | Chylomicron, VLDL, IDL, HDL | Ligand for remnant receptors (LDLR, LRP, proteoglycans); mediates hepatic uptake of CM remnants and IDL |
| Apo(a) | 187,000-662,000 | 6 | Lp(a) | Unknown; structurally homologous to plasminogen |
- Tietz Textbook of Laboratory Medicine, 7th ed., Table 36.4
ApoA Family
ApoA-I
The most important HDL apolipoprotein and the most cardioprotective. Together with ApoA-II, it makes up ~90% of total HDL protein (A-I : A-II ratio ≈ 3:1). Key roles:
- Structural scaffold of HDL - the degree of ApoA-I twisting around the HDL surface modulates particle size
- Cofactor for LCAT (lecithin-cholesterol acyltransferase) - LCAT esterifies free cholesterol, which is essential for HDL maturation and remodeling
- Ligand for ABCA1 - the major membrane transporter that mediates cellular cholesterol efflux onto nascent HDL (the first step of reverse cholesterol transport)
- ApoA-I can be present in 1-5 copies per HDL particle
Clinical significance: ApoA-I is a stronger predictor of cardiovascular protection than HDL-cholesterol alone. Low ApoA-I is associated with premature CVD.
ApoA-II
The second most abundant HDL protein. Its exact role remains incompletely understood, but evidence suggests it:
- Inhibits hepatic lipase (HL)
- Delays lipolysis of large TG-rich lipoproteins by interfering with LPL
ApoA-IV
Synthesized in the intestine, secreted as a component of chylomicrons. Functions:
- Activates LCAT
- Facilitates release of ApoC-II from HDL/VLDL (promoting LPL action)
- Promotes intestinal lipid absorption
- Satiety signaling via a hypothalamic effect
ApoA-V
A relatively low-abundance apolipoprotein found on VLDL and HDL. It lowers plasma triglycerides by:
- Enhancing LPL function (by facilitating lipoprotein interaction with the enzyme via GPI-anchored HDL-binding protein 1)
- Reducing VLDL secretion from the liver
Multiple polymorphisms of APOA5 are associated with hypertriglyceridemia.
ApoB Family
ApoB-100
The largest known apolipoprotein (>4,500 amino acids, ~513 kDa). Uniquely, it contains both amphipathic helices AND beta-sheets - this high lipid affinity means it is permanently embedded in its lipoprotein and cannot transfer between particles.
- Made exclusively in the liver
- Present on VLDL (secreted), IDL (transition), LDL (end-product), and Lp(a)
- The essential ligand for the LDL receptor (LDLR) - mutations in APOB that reduce LDLR binding cause familial hypercholesterolemia
- One molecule of ApoB-100 per LDL particle - so ApoB concentration is a direct measure of LDL particle number, often a better CVD risk marker than LDL-C
ApoB-48
Produced exclusively by the intestine - it is literally the N-terminal 48% of apoB-100, generated by RNA editing of the APOB transcript (the APOBEC enzyme inserts a premature stop codon). Key facts:
- Lacks the C-terminal LDLR-binding domain of apoB-100, so chylomicron remnants are cleared via apoE-mediated receptor pathways (LRP, LDLR), not directly via apoB-48
- One molecule per chylomicron
- Essential for chylomicron assembly and secretion of dietary lipid from the intestine
ApoC Family (Small Exchangeable Apolipoproteins)
All three ApoC proteins rapidly exchange between lipoproteins and are found in varying proportions on chylomicrons, VLDL, and HDL.
ApoC-I
- Activates LCAT (similar to ApoA-I)
- Inhibits clearance of chylomicrons from circulation (by blocking apoE-receptor interactions)
- Also inhibits cholesteryl ester transfer protein (CETP)
ApoC-II
- Essential cofactor for lipoprotein lipase (LPL) - without ApoC-II, LPL cannot efficiently hydrolyze triglycerides from TG-rich lipoproteins
- ApoC-II deficiency → familial LPL deficiency phenotype (Type I hyperlipoproteinemia) with massive hypertriglyceridemia and chylomicronemia
- Delivered to chylomicrons and VLDL from HDL once the particle enters circulation
ApoC-III
- Inhibits LPL (opposing ApoC-II)
- Inhibits hepatic uptake of remnant particles (blocking apoE-receptor interaction)
- Net effect: raises plasma TG levels
- Elevated ApoC-III is an independent cardiovascular risk factor
- Loss-of-function mutations in APOC3 are associated with very low TG levels and protection from CVD - this has driven interest in ApoC-III as a therapeutic target (e.g., antisense oligonucleotides like volanesorsen)
ApoE
A 34 kDa glycoprotein (299 amino acids), synthesized primarily by the liver but also locally in brain (astrocytes), macrophages, and other tissues.
Functions:
- Ligand for multiple receptors: LDLR, LRP (LDL-receptor-related protein), heparan sulfate proteoglycans
- Mediates hepatic clearance of chylomicron remnants and IDL
- Facilitates lipoprotein uptake across multiple receptor types
ApoE Isoforms: E2, E3, E4
Three common isoforms determined by amino acid substitutions at positions 112 and 158:
| Isoform | Position 112 | Position 158 | LDLR Binding | Effect on LDL-C |
|---|---|---|---|---|
| E2 | Cys | Cys | Reduced (~1% of E3) | Lowest LDL-C |
| E3 | Cys | Arg | Normal (reference) | Normal |
| E4 | Arg | Arg | Increased clearance but downregulates LDLR | Highest LDL-C |
Clinical implications of apoE genotype:
- ApoE2: Protective against atherosclerosis due to lower LDL-C; but when combined with other factors, can cause Type III hyperlipoproteinemia (dysbetalipoproteinemia) due to accumulation of remnant lipoproteins (because E2 binds LDLR poorly)
- ApoE3: Most common allele (~77% of population); reference phenotype
- ApoE4: Associated with higher LDL-C (increased cholesterol absorption + downregulation of LDLR); associated with statin hyporesponsiveness in some studies; strongly associated with late-onset Alzheimer disease and other neurodegenerative conditions (likely via effects on brain lipid metabolism)
Apo(a) and Lipoprotein(a) [Lp(a)]
Apo(a) is a unique apolipoprotein found only on Lp(a) particles. Key features:
- Structurally homologous to plasminogen (contains kringle domains) - this may explain the prothrombotic and pro-atherogenic properties of Lp(a)
- Linked to ApoB-100 by a single disulfide bond
- Highly variable molecular weight (187,000-662,000 Da) due to polymorphic number of kringle IV repeats
- Elevated Lp(a) is an independent, genetically determined risk factor for CVD and aortic stenosis
- Emerging therapeutic targets: PCSK9 inhibitors (modestly lower Lp(a)), and dedicated Lp(a)-lowering agents (pelacarsen, olpasiran) in trials
ApoD
- Structurally belongs to the lipocalin family (distinct from other apolipoproteins)
- Found on HDL
- Expressed highly in the brain, adrenal glands, and liver
- Proposed roles in transport of small lipophilic molecules; exact physiologic function not fully established
Lipoprotein-Apolipoprotein Associations at a Glance
| Lipoprotein | Key Apolipoproteins |
|---|---|
| Chylomicron | ApoB-48, ApoA-I, ApoA-IV, ApoC-I, ApoC-II, ApoC-III, ApoE |
| VLDL | ApoB-100, ApoC-I, ApoC-II, ApoC-III, ApoE |
| IDL | ApoB-100, ApoE |
| LDL | ApoB-100 (dominant; least protein diversity) |
| HDL | ApoA-I, ApoA-II, ApoA-IV, ApoC-I, ApoC-II, ApoC-III, ApoE (most protein diversity) |
| Lp(a) | ApoB-100 + Apo(a) |
Clinical Measurement
-
ApoA-I and ApoB are routinely measured by immunoturbidimetry or immunonephelometry in clinical labs (high concentration makes these methods feasible)
-
ApoC-I, ApoC-II (lower concentrations) require more sensitive methods such as ELISA
-
ApoB/ApoA-I ratio is a stronger predictor of CVD risk than the total cholesterol/HDL ratio in some studies
-
ApoE genotyping is used in selected clinical scenarios (familial dyslipidemias, Alzheimer risk counseling)
-
International standardization of ApoA-I and ApoB measurement is led by the IFCC Committee on Apolipoproteins
-
Henry's Clinical Diagnosis and Management by Laboratory Methods; Tietz Textbook of Laboratory Medicine, 7th ed.
This is a shared conversation. Sign in to Orris to start your own chat.