Acute leukaemia
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Searching PubMed
"acute leukemia" AND treatment
Reading File
Loading Image
Loading Image
Loading Image
I now have comprehensive, well-sourced information to compile the full answer on acute leukaemia.
Acute Leukaemia
Definition and Overview
Acute leukaemia (AL) is a rapidly progressing clonal malignancy of haematopoietic precursor cells in which immature blast cells proliferate uncontrollably in the bone marrow, suppress normal haematopoiesis, and spill into the peripheral blood and other organs. The defining criterion is ≥20% blasts in the marrow or peripheral blood (WHO classification). Without treatment, the course is rapidly fatal within weeks to months.
The two main categories are:
- Acute Myeloid Leukaemia (AML) - arising from myeloid precursors
- Acute Lymphoblastic Leukaemia (ALL) - arising from lymphoid precursors (B or T cell)
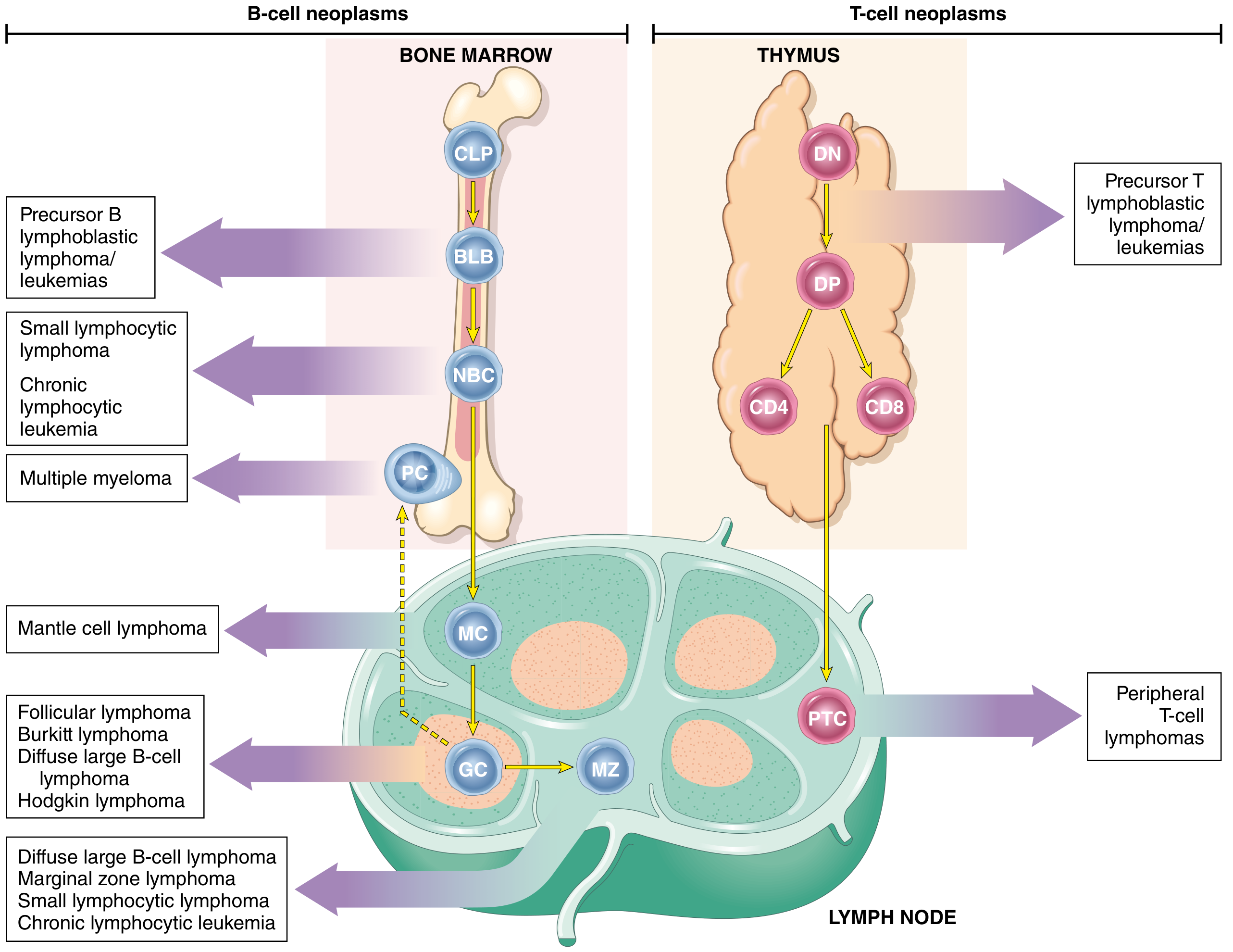
Fig: Origin of lymphoid and myeloid neoplasms - stages of B- and T-cell differentiation from which specific acute leukaemias emerge (Robbins, Cotran & Kumar - Pathologic Basis of Disease)
Epidemiology
AML:
- Most common acute leukaemia in adults
- Median age at diagnosis: ~60 years
- Incidence: ~10/100,000/year in those >60 years
- Most common form of acute leukaemia in the first months of life; accounts for ~1/3 of childhood acute leukaemias
ALL:
- Most common cancer of children (~2,500 new cases/year in the US)
- Peak incidence of B-ALL at age ~3 years
- T-ALL peaks in adolescence (adolescent males, thymic involvement)
- Hispanic/Latino children have the highest ALL incidence of any ethnic group
- Also occurs less commonly in adults
Risk factors for AML: prior radiation, cytotoxic chemotherapy, benzene, smoking; most cases have no identifiable cause.
- Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 743
Classification
WHO Classification Approach for AML
AML classification uses a multilayered approach recognizing:
- Recurrent acquired cytogenetic abnormalities
- History of predisposing factors (prior cytotoxic therapy)
- Association with MDS/related conditions
- Morphologic stratification for the remainder
The FAB classification (French-American-British, 1976) subdivided AML into M0-M7 based on morphology and cytochemistry. This has been largely superseded by the WHO classification but the basic morphologic tenets remain.
Key AML subtypes with recurrent cytogenetics:
| Cytogenetic Abnormality | Key Features | Prognosis |
|---|---|---|
| t(8;21); RUNX1::RUNX1T1 | Granulocytic maturation, Auer rods | Favorable |
| inv(16) or t(16;16); CBFB::MYH11 | Myelomonocytic + eosinophilia | Favorable |
| t(15;17); PML::RARA | APL - faggot cells, Auer rods, DIC | Favorable (with ATRA) |
| t(9;11); KMT2A::MLLT3 | Monocytic features | Intermediate |
| inv(3) or t(3;3); GATA2/MECOM | Multilineage dysplasia, thrombocytosis | Adverse |
| Complex karyotype / monosomal | - | Adverse |
| -5, -7, del(5q), del(7q) | Secondary AML, older patients | Adverse |
Key ALL immunophenotypic groups:
-
B-ALL (75%): Pro-B (CD19+, CD22+, CD10-); Common ALL (CD10/CALLA+, 50-60%); Pre-B (cytoplasmic Ig, ~10%); Mature B (surface Ig, <5%)
-
T-ALL (25%): Early T-precursor (CD7+, CD1a-, CD3-); Thymic (CD1a+); Mature (surface CD3+)
-
Goldman-Cecil Medicine, p. 1923
Pathogenesis
AML
AML cells carry an average of 10-15 mutations per cell. Key driver mutations include:
| Gene | Frequency | Prognostic Significance |
|---|---|---|
| NPM1 | ~30% | Favorable (if FLT3-ITD negative) |
| FLT3-ITD | ~25% of AML | Adverse (especially high allelic ratio) |
| FLT3-TKD | ~10% | Intermediate/adverse |
| CEBPA (biallelic) | 4-15% | Favorable |
| IDH1/IDH2 | Variable | Targetable; intermediate |
| DNMT3A | Common | Adverse |
| TP53 | - | Strongly adverse |
| RAS, RUNX1, TET2, ASXL1 | - | Variable/adverse |
Mutations in splicing factors (SRSF2, SF3B1, U2AF1, etc.) are almost exclusively found in secondary AML.
ALL
-
~90% of ALLs have numerical or structural chromosomal changes
-
T-ALL: NOTCH1 mutations in 50-70% (NOTCH1 is essential for T-cell development)
-
B-ALL: mutations in PAX5 (30%), IKZF1 (25%), and translocations involving ETV6, RUNX1, BCR::ABL1, KMT2A, PBX1
-
Philadelphia chromosome t(9;22): present in ~5% of childhood ALL but ~25% of adult ALL - constitutively activates ABL1 kinase (190 kD BCR-ABL1 in ALL vs 210 kD in CML)
-
Hyperdiploidy (>50 chromosomes): better prognosis in B-ALL
-
Hypodiploidy: worse prognosis in B-ALL
-
Ph-like ALL: 20-25% of adult ALLs; lacks BCR-ABL1 but has kinase-activating mutations (CRLF2, JAK2) - poor prognosis
-
Goldman-Cecil Medicine, p. 1922; Robbins & Kumar Pathologic Basis of Disease, p. 556-557
Clinical Manifestations
Signs and symptoms develop rapidly over weeks to months, resulting from marrow replacement and organ infiltration:
From marrow failure:
- Anaemia: fatigue, pallor, headache, exertional dyspnoea, angina
- Thrombocytopenia: petechiae, ecchymoses, bleeding gums, epistaxis, haemorrhage (clinically evident bleeding in ~1/3 at diagnosis)
- Neutropenia: infections (bacterial) - significant/life-threatening in ~1/3 of AML, slightly fewer in ALL
From organ infiltration (more prominent in ALL):
- Lymphadenopathy, hepatosplenomegaly (common in ALL)
- Mediastinal mass - characteristic of T-ALL
- Bone pain - common in children with ALL (periosteal infiltration)
- Leukemia cutis - raised, non-pruritic rash from skin infiltration
- CNS involvement - headache, nausea, cranial nerve palsies (leukemic meningitis)
- Gum hypertrophy - especially in monocytic AML subtypes
Metabolic complications:
-
Hyperuricaemia (esp. ALL)
-
Elevated LDH
-
DIC - hallmark of APL (acute promyelocytic leukaemia, AML M3)
-
Tumour lysis syndrome: hypocalcaemia, hyperkalaemia, hyperphosphataemia, hyperuricaemia, renal insufficiency
-
Goldman-Cecil Medicine, p. 1921-1922
Diagnosis
Peripheral blood:
- Anaemia and thrombocytopenia are nearly universal
- ~25% have platelets <20,000/µL
- WBC is variable: ~25% have WBC >50,000/µL (hyperleukocytosis), ~50% between 5,000-50,000, ~25% have low WBC (<5,000)
- Blasts usually present in peripheral blood
Bone marrow:
- Aspiration (± biopsy from posterior iliac crest) is the definitive diagnostic step
- Usually hypercellular with 20-100% blasts
- Diagnosis requires ≥20% blasts in marrow or blood
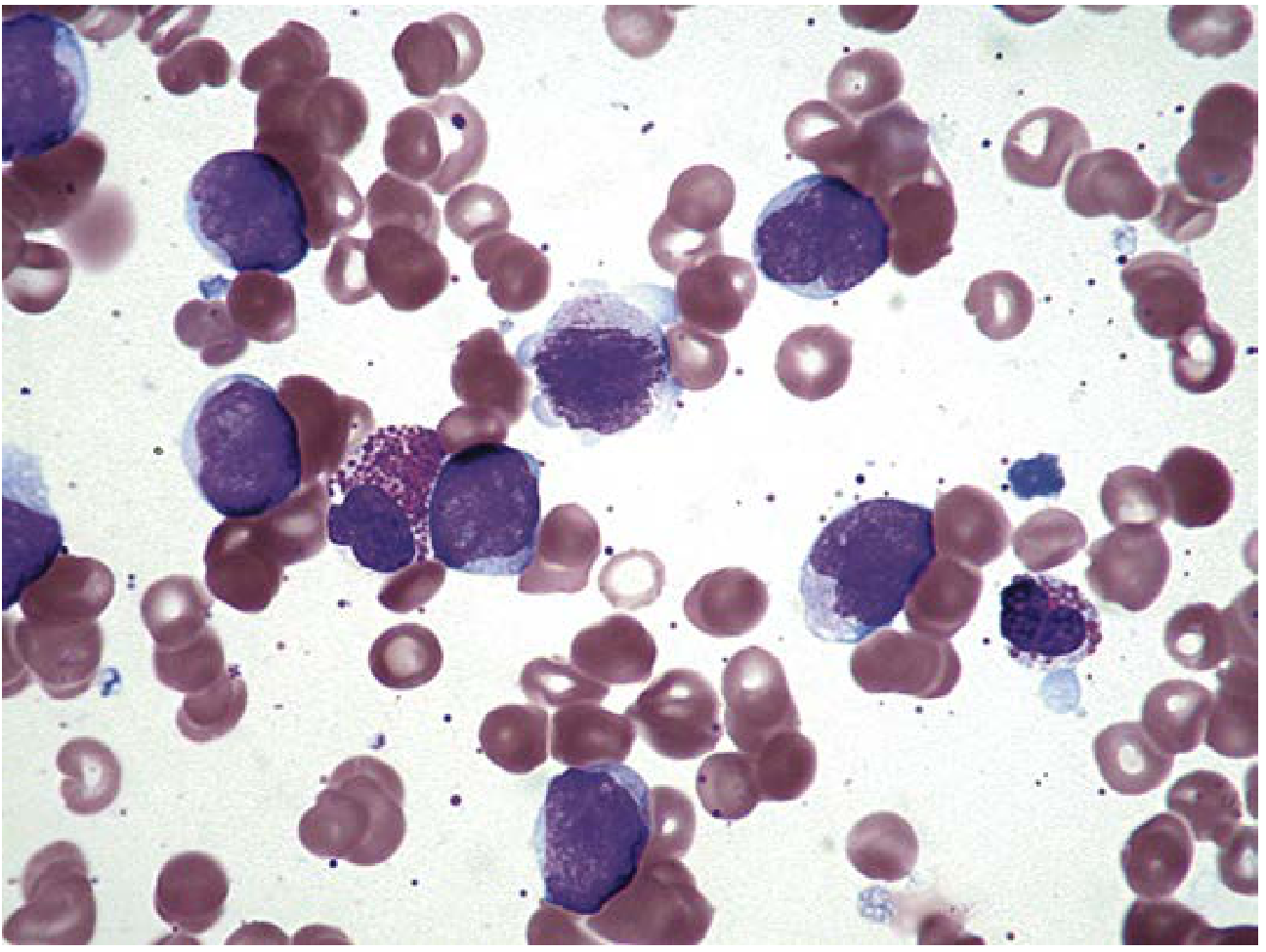
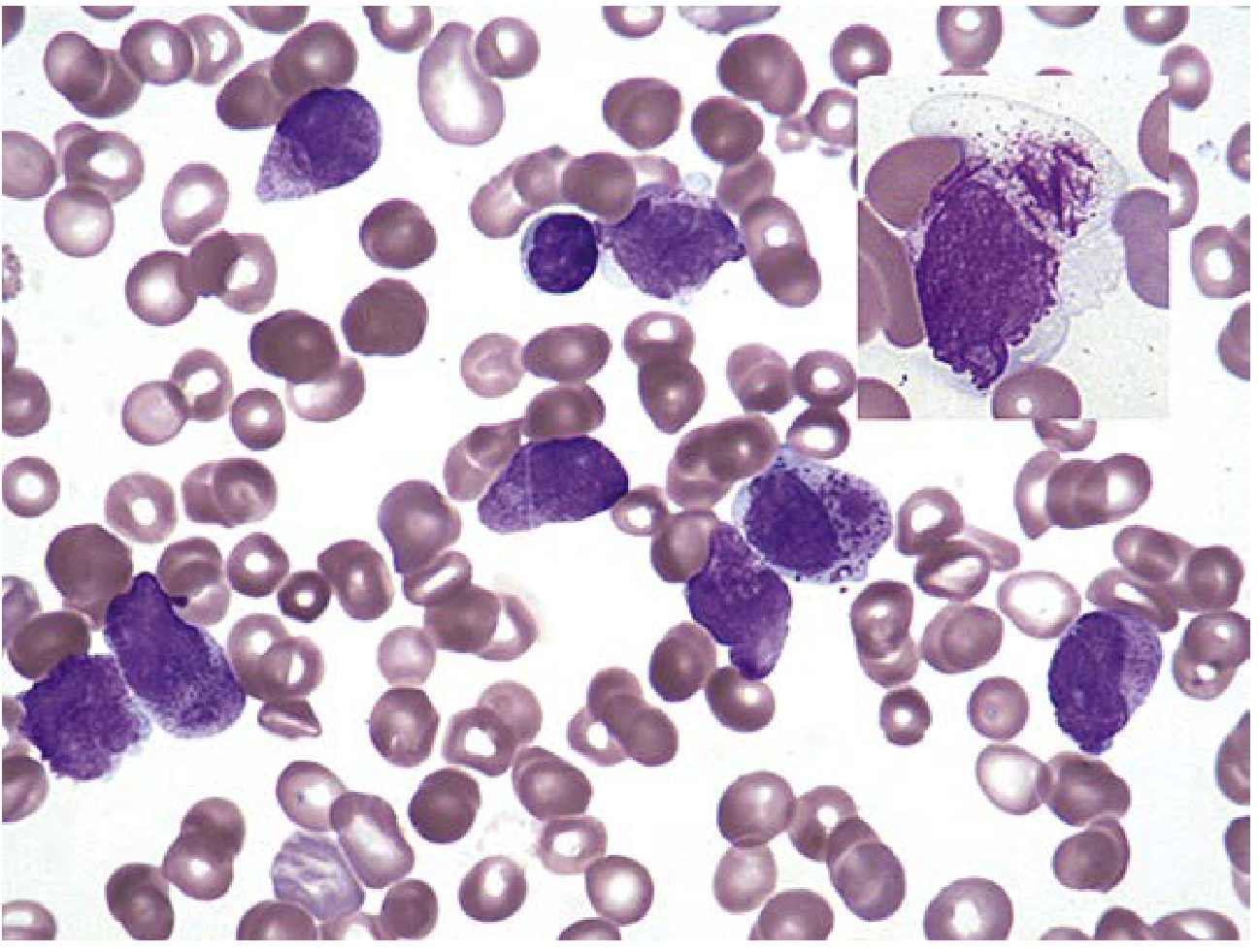
Immunophenotyping (flow cytometry):
- Identifies blast population (CD45 dim, low side scatter; CD34, CD117, CD133, TdT)
- Assigns lineage (myeloid vs lymphoid) and subtype
- Key myeloid markers: MPO, CD13, CD33, CD117, CD64
- Key B-lymphoid markers: CD19, CD22, CD10 (CALLA), CD79a
- Key T-lymphoid markers: CD3, CD7, CD1a, CD4, CD8
- Used for minimal residual disease (MRD) monitoring post-therapy
Additional workup:
- Cytogenetics (conventional karyotype)
- Molecular studies: FISH, PCR, next-generation sequencing (FLT3, NPM1, CEBPA, IDH1/2, TP53, etc.)
- Coagulation studies (PT, PTT, fibrinogen - screen for DIC, essential in APL)
- Lumbar puncture: recommended in ALL; not routine in AML unless CNS symptoms present
- LFTs, renal function, uric acid, LDH, urine electrolytes
Differential diagnosis:
-
Aplastic anaemia (hypocellular marrow, no blasts)
-
Myelodysplastic syndrome (<20% blasts)
-
Leukemoid reaction (blasts rarely reach 20%)
-
Infectious mononucleosis (mimics ALL)
-
Marrow infiltration by other small round cell tumours
-
Goldman-Cecil Medicine, p. 1922-1923; Henry's Clinical Diagnosis and Management, p. 786-787
Treatment
Acute Myeloid Leukaemia
Induction chemotherapy ("7+3"):
- Standard: cytarabine (continuous infusion x7 days) + anthracycline (daunorubicin or idarubicin x3 days)
- Goal: achieve complete remission (CR) - defined as <5% blasts in marrow, recovery of normal blood counts
APL - special treatment:
- All-trans retinoic acid (ATRA) + arsenic trioxide (ATO) is now standard first-line therapy
- This has transformed APL from the most lethal to the most curable subtype
- Beware of differentiation syndrome (dyspnoea, fever, pulmonary infiltrates) during treatment
- DIC must be managed aggressively with FFP, cryoprecipitate, platelet transfusions
Targeted therapy (mutation-specific):
- FLT3 inhibitors: midostaurin (added to induction for FLT3-mutated AML), gilteritinib (relapsed/refractory)
- IDH1 inhibitors: ivosidenib
- IDH2 inhibitors: enasidenib
- BCL-2 inhibitor: venetoclax (combined with hypomethylating agents - azacitidine/decitabine) - particularly for older/unfit patients
Post-remission/consolidation:
- Favorable-risk AML (t(8;21), inv(16), NPM1 mutated without FLT3-ITD): high-dose cytarabine consolidation
- Intermediate/adverse-risk AML: allogeneic haematopoietic cell transplantation (allo-HCT) in first CR
- Allo-HCT in first CR achieves disease-free survival of ~55-60%; meta-analyses confirm survival advantage over chemotherapy for adverse/intermediate-risk AML
Acute Lymphoblastic Leukaemia
Treatment phases in ALL:
- Induction (4-6 weeks): vincristine, glucocorticoids, asparaginase ± anthracycline; for Ph+ ALL, add tyrosine kinase inhibitor (imatinib/dasatinib/ponatinib)
- CNS prophylaxis: intrathecal methotrexate (±cytarabine); cranial radiation now largely replaced by intrathecal chemotherapy to reduce neurotoxicity
- Consolidation/intensification
- Maintenance: mercaptopurine, methotrexate for 2-3 years (critical in ALL to prevent relapse)
Key targeted agents in ALL:
- Imatinib/dasatinib/ponatinib for Ph+ ALL (BCR-ABL1)
- Blinatumomab (bispecific T-cell engager, CD19/CD3): used to achieve MRD-negative state before transplant
- Inotuzumab ozogamicin (anti-CD22 antibody-drug conjugate): relapsed/refractory B-ALL
- CAR-T cell therapy (tisagenlecleucel): relapsed/refractory B-ALL in children and young adults
Allo-HCT in ALL:
-
Adults in first CR: ~65% cure rate with transplant
-
Transplant in second CR: 40-50% cure rate
-
MRD negativity before transplant improves outcomes
-
Blinatumomab recommended to achieve MRD-negative state before transplantation
-
Harrison's Principles of Internal Medicine 22E (2025), p. 1522-1524; Goldman-Cecil Medicine, p. 1923-1925
Prognosis
AML prognostic risk groups (ELN 2022 framework):
| Risk | Features | 5-year OS |
|---|---|---|
| Favorable | t(8;21), inv(16)/t(16;16), NPM1 mut (FLT3-ITD-neg), biallelic CEBPA | ~50-60% |
| Intermediate | NPM1 mut + FLT3-ITD, t(9;11), normal karyotype without mutations | ~30-40% |
| Adverse | t(6;9), inv(3), TP53 mut, complex karyotype, monosomal karyotype, secondary AML | <20% |
ALL prognostic factors:
- Age (older = worse), WBC at presentation (high = worse)
- Ph+ ALL and Ph-like ALL: adverse (now partially overcome by TKI therapy)
- t(12;21) ETV6::RUNX1: favorable (25% of childhood B-ALL)
- Hyperdiploidy: favorable in B-ALL
- CNS involvement at diagnosis: adverse
- MRD status after induction: the most powerful prognostic factor
Overall: With modern therapy, ~40-50% of younger AML patients and ~50-70% of children with ALL achieve long-term remission/cure. Older patients (>60-70 years) with AML have substantially worse outcomes due to both disease biology and treatment intolerance.
Supportive Care
- Hyperuricaemia prophylaxis: allopurinol or rasburicase (especially before initiating treatment)
- Tumour lysis syndrome monitoring and management
- Broad-spectrum antibiotics for febrile neutropenia (empiric antipseudomonal coverage)
- Antifungal prophylaxis during prolonged neutropenia
- Platelet transfusions (threshold typically <10,000/µL or <20,000/µL with bleeding)
- Red cell transfusions for symptomatic anaemia
- G-CSF may shorten duration of neutropenia post-chemotherapy
Sources:
- Goldman-Cecil Medicine International Edition, Chapters 168/169
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 13
- Henry's Clinical Diagnosis and Management by Laboratory Methods, Chapter 34/35
- Harrison's Principles of Internal Medicine 22E (2025)
- Recent systematic review: Comparative efficacy in low-intensity AML treatment (Li et al., 2025, PMID 40229815)
Differentiate between all and aml
From the detailed information already gathered in our previous session, here is a comprehensive differentiation between ALL and AML:
ALL vs AML - Complete Differentiation
1. Basic Identity
| Feature | ALL (Acute Lymphoblastic Leukaemia) | AML (Acute Myeloid Leukaemia) |
|---|---|---|
| Cell of origin | Lymphoid precursor (B or T lymphoblast) | Myeloid precursor (myeloblast, monoblast, erythroblast, megakaryoblast) |
| Alternate name | Acute lymphocytic leukaemia | Acute myelogenous leukaemia |
| Blast type | Lymphoblasts | Myeloblasts (± Auer rods) |
2. Epidemiology
| Feature | ALL | AML |
|---|---|---|
| Primary age group | Children (peak at age 3 years) | Adults (median age ~60 years) |
| Most common cancer in | Children (<15 years) | Older adults |
| Gender | Slightly more common in boys | No strong gender predominance |
| Ethnic predisposition | Hispanic/Latino children - highest incidence | None well-defined |
| In infancy | Less common than AML in first months | Most common AL in first few months of life |
3. Morphology
| Feature | ALL | AML |
|---|---|---|
| Blast morphology | Small-medium lymphoblasts, scant cytoplasm, condensed chromatin, inconspicuous nucleoli | Large myeloblasts, abundant cytoplasm, fine chromatin, prominent nucleoli (3-5), cytoplasmic granules |
| Auer rods | Absent | Present (pathognomonic when seen) |
| Special variant | - | APL (M3): faggot cells (multiple Auer rod bundles) |
| Cytochemistry - MPO | Negative | Positive (key distinguishing stain) |
| Sudan Black B (SBB) | Negative | Positive |
| PAS stain | Positive (chunky block positivity) | Negative or diffuse weak positivity |
| Non-specific esterase (NSE) | Negative | Positive in monocytic subtypes |
4. Immunophenotype
| Marker | ALL | AML |
|---|---|---|
| TdT (terminal deoxynucleotidyl transferase) | Positive (hallmark) | Negative (except some AML-M0) |
| CD34 | Often positive | Often positive |
| CD10 (CALLA) | Positive in common B-ALL | Negative |
| CD19, CD22, CD79a | Positive (B-ALL) | Negative |
| CD3, CD7 | Positive (T-ALL) | Negative |
| CD13, CD33, CD117 | Negative | Positive |
| MPO (cytoplasmic) | Negative | Positive |
| CD64 | Negative | Positive (mature myeloid) |
| CD41, CD61 | Negative | Positive in megakaryoblastic AML |
5. Genetics and Molecular Biology
| Feature | ALL | AML |
|---|---|---|
| Most common translocation in children | t(12;21) ETV6::RUNX1 - 25%, favorable | t(8;21) RUNX1::RUNX1T1 - favorable |
| Philadelphia chromosome t(9;22) | Present in ~5% children, ~25% adults - ADVERSE | Present in rare de novo AML (<1%), very adverse |
| Most common adult ALL mutation | t(9;22) BCR-ABL1 (190 kD protein) | FLT3-ITD (~25-30%), NPM1 (~30%) |
| NPM1 mutations | Rare | ~30% - favorable (without FLT3-ITD) |
| FLT3 mutations | Rare | ~30-35% - adverse (especially high allelic ratio) |
| NOTCH1 mutations | T-ALL: 50-70% | Absent |
| PAX5/IKZF1 | B-ALL: 30% and 25% respectively | Absent |
| APL-specific | Absent | t(15;17) PML::RARA - unique to APL |
| KMT2A (MLL) rearrangements | Infant ALL (very adverse) | t(9;11) - intermediate |
| Hyperdiploidy (>50 chr) | B-ALL - favorable | Not applicable |
| Hypodiploidy | B-ALL - adverse | Not applicable |
| Ph-like ALL | 20-25% of adult ALL - adverse | Not applicable |
6. Clinical Features
| Feature | ALL | AML |
|---|---|---|
| Lymphadenopathy | Prominent (common at diagnosis) | Mild or absent |
| Hepatosplenomegaly | Common and prominent | Less pronounced |
| Mediastinal mass | T-ALL - frequent (thymic involvement) | Rare |
| Bone pain | Common, especially in children (periosteal infiltration) | Less common |
| CNS involvement | More frequent (headache, cranial nerve palsies) | Less common |
| Gum hypertrophy | Absent | Monocytic AML subtypes |
| Skin infiltration (leukemia cutis) | Less common | Monocytic AML |
| DIC | Uncommon | APL (AML M3) - hallmark |
| Infections at presentation | Common (~1/3), slightly fewer than AML | ~1/3, more pronounced |
| Hyperuricaemia | More common (rapid cell turnover) | Present but less striking |
7. Laboratory Findings
| Finding | ALL | AML |
|---|---|---|
| Anaemia | Universal | Universal |
| Thrombocytopenia | Universal | Universal |
| WBC | Variable; can be very high | Variable |
| LDH | Markedly elevated | Elevated |
| Uric acid | Often very high | Elevated |
| Coagulation (DIC) | Usually normal | Abnormal in APL (low fibrinogen, prolonged PT/PTT) |
| Serum lysozyme | Normal | Elevated in monocytic AML |
| CSF involvement at diagnosis | More frequent | Less frequent |
8. Treatment
| Treatment Aspect | ALL | AML |
|---|---|---|
| Induction backbone | Vincristine + steroids + asparaginase ± anthracycline | "7+3": cytarabine x7d + anthracycline x3d |
| Phases of therapy | Induction → Consolidation → Maintenance (2-3 years) | Induction → Consolidation (no prolonged maintenance) |
| Maintenance therapy | Essential (mercaptopurine + methotrexate) | Not standard (except APL) |
| CNS prophylaxis | Mandatory (intrathecal MTX ± cytarabine) | Only if CNS symptoms (not routine) |
| APL treatment | Not applicable | ATRA + arsenic trioxide (highly specific) |
| Targeted therapy | TKIs (imatinib/dasatinib) for Ph+ ALL; blinatumomab; inotuzumab; CAR-T | FLT3 inhibitors (midostaurin, gilteritinib); IDH1/2 inhibitors; venetoclax |
| Lumbar puncture at diagnosis | Recommended in all patients | Only if CNS symptoms |
| Allo-HCT in first CR | High-risk and Ph+ ALL | Intermediate/adverse-risk AML |
9. Prognosis
| Feature | ALL | AML |
|---|---|---|
| Children | Excellent: ~90% cure rate in standard-risk | ~60-70% with intensive therapy |
| Adults | ~40-50% (worse than children) | ~30-40% in younger adults; <20% in elderly |
| Best prognostic subtype | Childhood B-ALL with t(12;21) or hyperdiploidy | APL with t(15;17) - >90% cure rate with ATRA+ATO |
| Worst prognostic subtype | Ph+ ALL (partially overcome with TKIs); infant KMT2A-rearranged | Complex karyotype, monosomal karyotype, TP53 mutation |
| MRD monitoring | Critical - MRD negativity predicts sustained remission | Important - used to guide transplant decisions |
10. Key "One-Line" Distinguishing Points
| Feature | Favors ALL | Favors AML |
|---|---|---|
| Child under 15 | ✓ | |
| Adult over 60 | ✓ | |
| TdT positive | ✓ | |
| MPO positive / Auer rods | ✓ | |
| Mediastinal mass | ✓ (T-ALL) | |
| DIC at presentation | ✓ (APL) | |
| t(9;22) in adult | ✓ (more common in ALL) | Less common |
| Prominent lymphadenopathy | ✓ | |
| Gum hypertrophy | ✓ (monocytic) | |
| 2-3 year maintenance therapy needed | ✓ | |
| ATRA responsive | ✓ (APL only) |
Sources:
- Goldman-Cecil Medicine International Edition, Chapters 168/169
- Robbins, Cotran & Kumar Pathologic Basis of Disease, Chapter 13
- Henry's Clinical Diagnosis and Management by Laboratory Methods, Chapters 34/35
- Harrison's Principles of Internal Medicine 22E (2025)
Types of fever
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
I now have rich textbook material to compile a comprehensive answer. Here is the full breakdown:
Types of Fever
Fever is classified based on its temperature pattern over time (duration, height, fluctuation, and periodicity). Understanding the pattern is diagnostically important as each pattern is characteristically associated with specific diseases.
Normal Body Temperature (Reference)
| Site | Normal Range |
|---|---|
| Oral | 36.4 - 37.4°C (97.5 - 99.3°F) |
| Axillary | ~1°F (0.6°C) lower than oral |
| Rectal | ~1°F higher than oral |
| Core (rectal/tympanic) | Up to 37.8°C (100°F) accepted as upper limit of normal |
Fever = core body temperature >38°C (100.4°F)
Classification by Pattern
1. Continuous (Sustained) Fever
- Temperature remains above normal and does not fluctuate more than 1°C (2°F) in 24 hours
- Temperature never returns to normal
- Sustained high plateau
Causes:
- Typhoid fever (classic: stepladder rise in week 1 → continuous plateau in week 2)
- Lobar pneumonia
- Urinary tract infection
- Rickettsial infections (typhus)
- Meningitis
"When the fever does not fluctuate for more than 1°C during 24 hours, but at no time touches the normal, it is described as continued."
- S. Das Manual on Clinical Surgery, p. 11
2. Remittent Fever
- Temperature daily fluctuations exceed 2°C but never touches normal between fluctuations
- Temperature remains elevated throughout
- Most common fever pattern seen in clinical practice
Causes:
- Infective endocarditis
- Brucellosis ("undulant fever" - classic remittent/undulating character)
- Typhoid (early stages)
- Viral infections
- Most bacterial infections
- Pel-Ebstein fever (a special remittent pattern - see below)
3. Intermittent Fever
- Temperature rises then falls back to normal or below normal each day
- There is a clear afebrile period between spikes
- The spike pattern gives diagnostic clues:
| Sub-type | Spike frequency | Plasmodium species |
|---|---|---|
| Quotidian | Daily (every 24 hours) | P. falciparum (early), P. knowlesi |
| Tertian | Every 48 hours (alternate days) | P. vivax, P. ovale (Benign tertian); P. falciparum (Malignant tertian) |
| Quartan | Every 72 hours (every 3rd day) | P. malariae |
| Double tertian | Twice daily | Mixed P. vivax infection |
Other causes of intermittent fever:
- Pyogenic abscesses
- Kala-azar (visceral leishmaniasis)
- Lymphoma
- Filariasis (periodic fever, often correlating with full/new moon)
"These regular fever patterns (quotidian, daily; tertian, every 2 days; quartan, every 3 days) are seldom seen today as patients receive prompt and effective antimalarial treatment."
- Harrison's Principles of Internal Medicine 22E (2025)
4. Hectic (Septic) Fever
- Also called "swinging fever" or "picket fence fever"
- Very wide daily swings in temperature (often >2°C), alternating between very high spikes and near/below normal
- Associated with rigors (chills, shivering) during temperature rise and drenching sweats during defervescence
- Essentially a severe form of remittent/intermittent fever
Causes:
- Pyaemia and septicaemia
- Deep-seated abscess (liver abscess, subphrenic abscess, renal abscess)
- Lateral sinus thrombosis (classic "picket fence" pattern - diurnal temperature spikes >39.4°C)
- Miliary tuberculosis
- Infective endocarditis
- P. falciparum malaria (in early phase before synchronization)
"The classic initial symptoms of lateral sinus thrombosis include a picket fence fever pattern (diurnal temperature spikes that exceed 103°F/39.4°C)."
- Cummings Otolaryngology Head and Neck Surgery
5. Relapsing (Recurrent) Fever
- Episodes of fever lasting several days, followed by an afebrile period of several days, then recurrence
- Pattern repeats multiple times
Causes:
- Relapsing fever (Borrelia recurrentis - louse-borne; Borrelia spp. - tick-borne)
- Malaria (recrudescence)
- Brucellosis (relapsing pattern)
- Rat-bite fever
- Hodgkin's lymphoma (Pel-Ebstein fever - see below)
6. Pel-Ebstein Fever
- A special relapsing pattern described in Hodgkin's disease
- Weeks of high fever alternating with weeks of normal/low temperature
- The cycle repeats regularly
- Classically described as "intermittent bouts of remittent fever"
Causes:
- Hodgkin's lymphoma (pathognomonic when present)
- Also reported in other lymphomas
"In Hodgkin's disease, intermittent bouts of remittent fever (Pel-Ebstein fever) is quite peculiar to this disease."
- S. Das Manual on Clinical Surgery, p. 114
7. Biphasic (Saddle-back) Fever
- Two distinct fever peaks separated by a brief afebrile period ("saddle" shape on chart)
- First spike: viraemia phase
- Afebrile dip: brief remission
- Second spike: immune/end-organ phase
Causes:
- Dengue fever (classic biphasic or "saddle back" pattern)
- Yellow fever
- Poliomyelitis (minor illness → afebrile → major paralytic illness)
- Leptospirosis (two phases)
- Colorado tick fever
8. Inverse (Typhoid) Fever
- Temperature is higher in the morning than in the evening
- This is the reverse of normal diurnal variation (normally temperature peaks in the evening)
Causes:
- Tuberculosis (especially miliary TB)
- Typhoid (occasionally)
- Typhus
9. Step-ladder (Typhoid) Fever
- Temperature rises in a step-wise pattern over several days, each peak higher than the last
- Classic in the first week of typhoid fever
Cause:
- Typhoid fever (Salmonella Typhi)
Summary Table
| Fever Type | Pattern | Key Causes |
|---|---|---|
| Continuous | Always elevated; <1°C fluctuation | Typhoid (plateau), lobar pneumonia, typhus |
| Remittent | Always elevated; >2°C fluctuation, never normal | Most bacterial infections, brucellosis, endocarditis |
| Intermittent | Spikes to normal between episodes | Malaria, abscess, kala-azar |
| - Quotidian | Daily spike | P. falciparum, P. knowlesi |
| - Tertian | Every 48 hrs | P. vivax (benign), P. falciparum (malignant) |
| - Quartan | Every 72 hrs | P. malariae |
| Hectic/Septic | Wild swings, rigors + sweats | Septicaemia, pyaemia, deep abscesses |
| Relapsing | Days of fever → afebrile → recurrence | Borrelia (relapsing fever), malaria recrudescence |
| Pel-Ebstein | Weeks of fever → weeks afebrile | Hodgkin's lymphoma |
| Biphasic/Saddle-back | Two peaks with dip in between | Dengue, leptospirosis, yellow fever |
| Step-ladder | Stepwise rise each day | Typhoid (week 1) |
| Inverse | Higher in morning | TB, typhoid |
Special Named Fevers by Disease
| Disease | Characteristic Fever Pattern |
|---|---|
| Typhoid | Stepladder rise (week 1) → Continuous plateau (week 2) → Gradual lysis (week 4) |
| Malaria (P. vivax) | Tertian (48-hr) intermittent with cold → hot → wet stages |
| Malaria (P. falciparum) | Initially quotidian, then tertian (malignant tertian), hectic if unsynchronised |
| Malaria (P. malariae) | Quartan (72-hr) intermittent |
| Brucellosis | Undulant (remittent/relapsing) with evening rise |
| Hodgkin's lymphoma | Pel-Ebstein (weeks of fever, weeks afebrile) |
| Dengue | Saddle-back biphasic |
| Infective endocarditis | Remittent or hectic |
| Septicaemia/pyaemia | Hectic with rigors and sweats |
| Tuberculosis | Evening rise; inverse diurnal pattern in miliary TB |
| Filariasis | Periodic (often related to lunar cycle) |
Sources:
- S. Das - A Manual on Clinical Surgery, 13th Edition, pp. 11, 114
- Harrison's Principles of Internal Medicine 22E (2025)
- Medical Microbiology 9e
- Sleisenger and Fordtran's Gastrointestinal and Liver Disease
- Cummings Otolaryngology Head and Neck Surgery
This is a shared conversation. Sign in to Orris to start your own chat.