Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND mechanism
Finding Sources
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND "pathophysiology"[tiab]
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
I now have thorough coverage across multiple authoritative textbooks. I have enough to write a comprehensive, well-structured answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
ARDS is a syndrome of noncardiogenic pulmonary edema driven by diffuse alveolar damage (DAD). Unlike cardiogenic edema (elevated hydrostatic pressure), ARDS arises from a breakdown of the alveolar-capillary barrier, allowing protein-rich exudate to flood the airspaces and causing refractory hypoxemia and decreased lung compliance. - Murray & Nadel's Textbook of Respiratory Medicine
1. Triggers and Initiating Insults
ARDS arises from either a direct (pulmonary) or indirect (extrapulmonary) insult:
| Direct (pulmonary) | Indirect (systemic) |
|---|---|
| Pneumonia (bacterial, viral, fungal) | Sepsis (most common cause overall) |
| Aspiration of gastric contents | Severe trauma / polytrauma |
| Inhalation injury | Pancreatitis |
| Pulmonary contusion | Transfusion (TRALI) |
| Drowning | Burns, shock |
2. Phase Structure of ARDS
The disease follows three overlapping, sequential phases:

3. The Exudative Phase (Days 0-7): Core Pathogenic Mechanism
This is the mechanistically richest phase and the one that defines ARDS.
3a. Alveolar-Capillary Barrier Disruption
The alveolar-capillary unit consists of the type I and type II alveolar epithelium on one side and the microvascular endothelium on the other. Under normal conditions these two cell layers maintain tight barrier function.
In ARDS, both layers are damaged simultaneously:
- Endothelial injury: inflammatory signals (e.g., LPS, TNF-alpha, IL-1beta) cause endothelial cell activation, retraction of tight junctions, and increased paracellular permeability. Loss of endothelial barrier integrity is both necessary and sufficient for ARDS development.
- Epithelial injury: Type I pneumocytes (which cover ~95% of alveolar surface) are particularly vulnerable. Their death occurs via necrosis, apoptosis, mechanical stretch, and coagulation-mediated pathways. Type II pneumocytes are more resistant but are also damaged.
The result is a high-permeability leak - protein-rich, exudative fluid (not hydrostatic transudate) floods the alveolar spaces. - Murray & Nadel's
3b. The Central Role of Neutrophils
Neutrophils are the key effector cells of ARDS:
- Recruitment: Alveolar macrophages sense the initial insult (PAMPs, DAMPs) and release chemokines including IL-8 (CXCL8), TNF-alpha, and IL-1beta, which recruit circulating neutrophils into the pulmonary vasculature and alveoli.
- Sequestration: Neutrophils become sequestered in pulmonary capillaries partly because their large diameter relative to capillary diameter forces them to deform, slowing transit; inflammatory signals further promote adhesion via endothelial ICAM-1 and E-selectin upregulation.
- Tissue destruction: Activated neutrophils release a destructive arsenal:
- Proteases (elastase, matrix metalloproteinases) - degrade extracellular matrix and surfactant protein A
- Reactive oxygen species (ROS) - directly damage epithelial and endothelial cell membranes
- Myeloperoxidase - generates hypochlorous acid
- Neutrophil extracellular traps (NETs) - also implicated in vascular injury
- These products together cause the epithelial necrosis and endothelial permeability increase that define DAD. - Robbins & Kumar Basic Pathology; Murray & Nadel's
3c. Macrophage-Driven Cytokine Storm
Alveolar macrophages (mφ) serve both as the initiators and amplifiers of lung injury:
- They release TNF-alpha, IL-1beta, IL-6, IL-8, platelet-activating factor (PAF), and complement fragments
- These cytokines perpetuate neutrophil recruitment and activate the coagulation cascade
- IL-6 in particular contributes to diffuse alveolar damage and multi-organ dysfunction - Schwartz's Principles of Surgery; Murray & Nadel's
3d. Coagulation and Fibrin Deposition
The inflammatory and coagulation pathways are tightly intertwined in ARDS:
- Endothelial injury exposes subendothelial collagen, activating the extrinsic coagulation cascade
- Alveolar macrophages express tissue factor, further driving fibrin generation
- Simultaneously, fibrinolysis is suppressed (elevated PAI-1 levels), shifting the balance toward clot formation
- Intravascular fibrin microthrombi form in pulmonary capillaries, contributing to pulmonary hypertension and dead space
- Intra-alveolar fibrin combines with cellular debris to form the characteristic hyaline membranes - Murray & Nadel's; Goldman-Cecil
3e. Surfactant Dysfunction
Type II pneumocytes produce and recycle surfactant. In ARDS:
- Damaged type II cells produce less surfactant
- Neutrophil elastase degrades surfactant protein A, a key component
- Plasma proteins leaking into the airspace inhibit surfactant function
- The ratio of large (active) to small (inactive) surfactant aggregates falls
- The net result is increased alveolar surface tension, predisposing to widespread alveolar collapse (atelectasis) and worsening V/Q mismatch - Murray & Nadel's
3f. Physiologic Consequences of the Exudative Phase
- Hypoxemia: from right-to-left shunt (perfused but non-ventilated flooded alveoli) and V/Q mismatch
- Decreased lung compliance: fluid-filled, collapsed alveoli are stiff
- Increased dead space: pulmonary microvascular thrombosis eliminates gas exchange in some regions
- Pulmonary hypertension: from hypoxic vasoconstriction, fibrin microthrombi, and vascular compression by PEEP
- Increased minute ventilation: respiratory compensation leads to rapid shallow breathing
4. The Proliferative Phase (Days 7-21)
The key features of this phase are:
- Type II pneumocyte hyperplasia: surviving type II cells proliferate to re-epithelialize denuded alveolar surfaces, then differentiate into type I cells
- Interstitial inflammation: fibroblasts infiltrate, beginning collagen deposition
- Hyaline membrane organization: fibrin matrices are remodeled
- Decreased neutrophils: the acute inflammatory burden diminishes
- Most patients show gradual improvement in oxygenation during this phase - Harrison's Principles
5. The Fibrotic Phase (Day 21+)
Only a minority of patients enter this phase. Characterized by:
- Progressive pulmonary fibrosis: collagen deposition in alveolar walls and interstitium
- Bullae formation, obliteration of pulmonary capillaries
- Sustained hypoxemia and ventilator dependence
- Fibroproliferation may begin very early - elevated N-terminal procollagen peptide III (a marker of collagen synthesis) can be detected in BAL fluid within 24 hours of ARDS onset - Murray & Nadel's
6. Ventilator-Induced Lung Injury (VILI) - A Secondary Mechanism
Mechanical ventilation itself perpetuates injury through two main mechanisms:
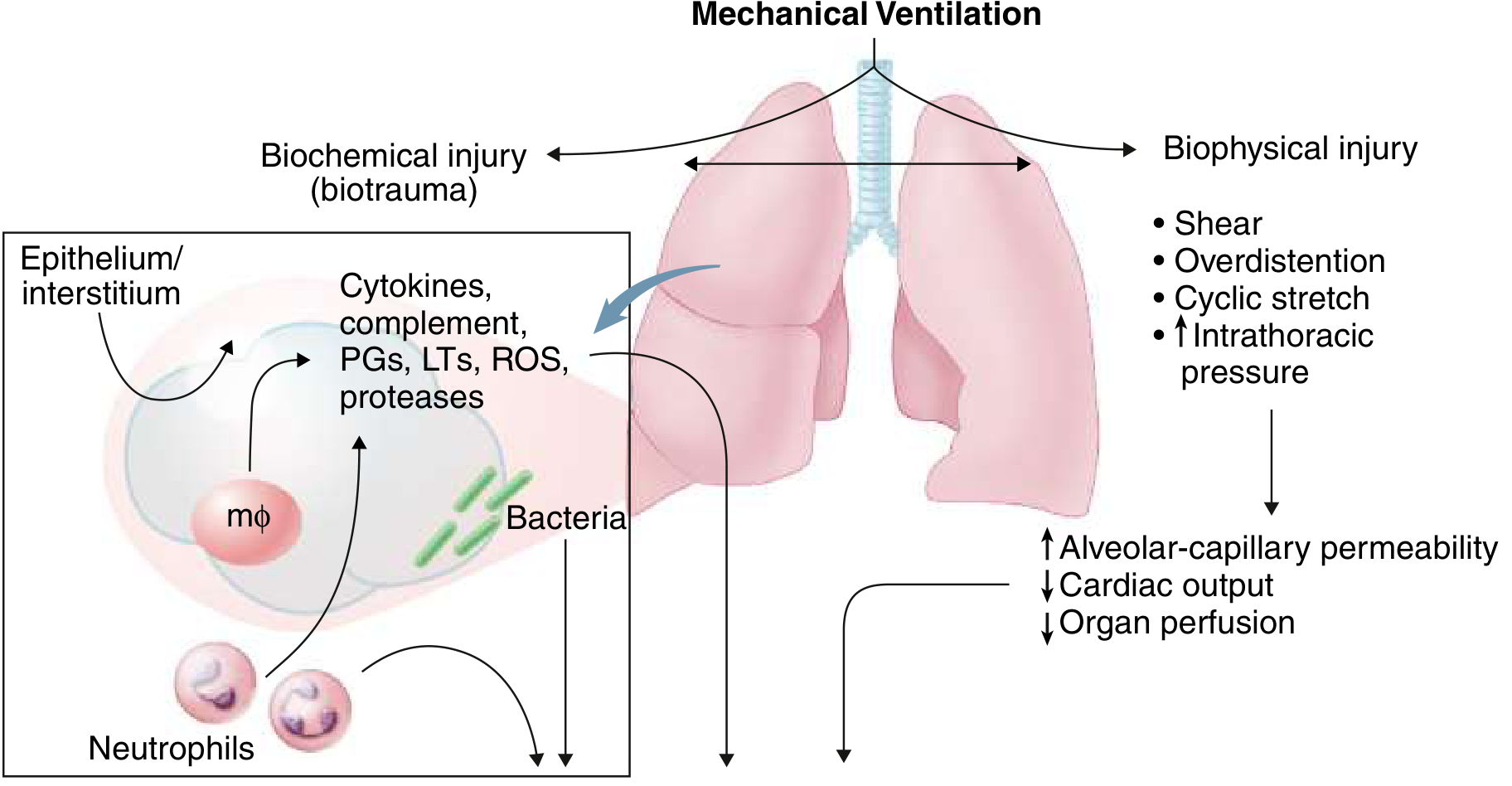
- Volutrauma: large tidal volumes overdistend non-injured lung units (ARDS creates heterogeneous disease, so a tidal volume meant for the whole lung goes preferentially to the "baby lung" of relatively normal alveoli)
- Atelectrauma: repetitive opening and closing of collapsed alveoli generates shear forces at the interface of collapsed and aerated tissue
- Biotrauma: mechanical stretch itself releases cytokines (IL-8, IL-6, TNF-alpha) from alveolar cells, creating a positive feedback loop that can injure distal organs
This is why the ARDSNet low tidal volume strategy (6 mL/kg predicted body weight) is the only intervention proven to reduce ARDS mortality. - Murray & Nadel's; Goldman-Cecil
7. Angiopoietin Signaling
A more recently appreciated pathway involves vascular stabilization proteins:
- Angiopoietin-1 (Ang1): enhances endothelial barrier integrity via the Tie2 receptor
- Angiopoietin-2 (Ang2): a competitive antagonist that destabilizes the endothelial barrier and promotes vascular leak
- In ARDS and sepsis, Ang2 levels are markedly elevated, contributing to sustained permeability
- Genetic variants in the Ang2 gene are associated with higher plasma Ang2 and increased ARDS risk - Murray & Nadel's
8. Genetic and Subphenotype Determinants
ARDS is not a single uniform entity. At least two biological subphenotypes have been described:
- Hyperinflammatory phenotype: high IL-8, IL-6, TNF receptor-1, low protein C - worse mortality, more vasopressor-dependent shock
- Hypo-inflammatory phenotype: milder inflammatory burden, better prognosis
These subphenotypes may respond differently to fluid management, PEEP strategies, and drug therapy - reflecting the underlying mechanistic heterogeneity. - Murray & Nadel's
Summary Diagram of Core Mechanism
Initiating insult (sepsis, pneumonia, trauma, aspiration)
↓
Alveolar macrophage activation
↓
Cytokine release (TNF-α, IL-1β, IL-6, IL-8, PAF)
↓
Neutrophil recruitment & sequestration in alveoli
↓
Neutrophil products: proteases, ROS, NETs
↓
Type I pneumocyte + endothelial cell injury/death
↓
Alveolar-capillary barrier breakdown
↓
Protein-rich exudate floods alveoli (DAD + hyaline membranes)
↓
Surfactant dysfunction → alveolar collapse
Coagulation activation → microthrombi → dead space
↓
Hypoxemia (shunt + V/Q mismatch) + decreased compliance
↓
RESPIRATORY FAILURE
Sources: Murray & Nadel's Textbook of Respiratory Medicine | Harrison's Principles of Internal Medicine, 22e | Robbins & Kumar Basic Pathology | Goldman-Cecil Medicine
This is a shared conversation. Sign in to Orris to start your own chat.