Billary atresia
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Searching Images
biliary atresia Kasai portoenterostomy pathology
Reading File
Loading Image
Here is a comprehensive overview of Biliary Atresia.
Biliary Atresia
Definition
Biliary atresia is a fibroproliferative obliteration of the extrahepatic biliary tree occurring within the first 3 months of life, resulting in complete obstruction of bile flow. It is the most frequent cause of death from liver disease in early childhood and accounts for ~50–60% of pediatric liver transplant referrals. — Robbins & Kumar Basic Pathology
Epidemiology
- Incidence: 1 in 8,000–18,000 live births in the USA
- Accounts for ~one-third of cases of neonatal cholestatic jaundice
- Slight female predominance
- Not inherited; discordant in dizygotic and monozygotic twins
- Non-Hispanic Black infants may carry higher risk
- Not associated with seasonality or time-space clustering
— Sleisenger & Fordtran's GI and Liver Disease; Schwartz's Principles of Surgery
Classification / Pathogenesis
Two major forms exist:
| Form | Frequency | Features |
|---|---|---|
| Fetal (embryonic) | ~20% of cases | Associated with other developmental anomalies — polysplenia, intestinal malrotation, preduodenal portal vein, congenital heart disease, interrupted IVC |
| Perinatal (acquired) | ~80% of cases | Normally formed biliary tree is injured/obstructed after birth; etiology unknown |
Proposed mechanisms include:
- Viral injury: CMV (56% of BA patients had elevated IFN-γ liver T-cells in response to CMV), rotavirus group C, reovirus type 3, EBV
- Immune dysregulation: autoimmunity, dysregulated T-regulatory cell function, coordinated activation of lymphocyte differentiation genes
- Genetic susceptibility: GPC1 (glypican-1, regulates Hedgehog signaling), ADD3, XPNPEP1 at locus 10q24.2; cfc1 gene mutations in biliary atresia-splenic malformation syndrome
- Toxin exposure: plant isoflavonoid ingestion linked to BA in livestock and zebrafish models
- Maternal microchimerism: expression of maternal antigens may trigger autoimmune bile duct destruction
— Sleisenger & Fordtran's; Schwartz's
Morphology / Pathology
- Inflammation and fibrosing stricture of the hepatic or common bile ducts
- Periductular inflammation may extend into intrahepatic ducts → progressive destruction
- Bile duct proliferation at portal plates (seen on trichrome staining) — a nonspecific but characteristic marker
- Intrahepatic bile ducts are NEVER dilated in biliary atresia (key distinguishing feature)
- If unrecognized/untreated: cirrhosis within 3–6 months of birth
Pattern of obstruction (surgically critical):
- ~10%: distal duct is patent; gallbladder may be visible — proximal ducts still atretic
- ~90%: obstruction at or above the porta hepatis — no patent bile ducts amenable to anastomosis
- When limited to common duct or hepatic ducts with patent intrahepatic branches → surgically correctable
— Robbins & Kumar; Schwartz's
Clinical Presentation
- Jaundice at birth or shortly thereafter (easily confused with physiologic neonatal jaundice, causing diagnostic delay)
- Acholic (pale gray) stools — hallmark of obstructed bile flow; stools may initially appear normal, becoming acholic with disease progression
- Dark urine (conjugated hyperbilirubinemia)
- Progressive failure to thrive
- If untreated: portal hypertension → splenomegaly, esophageal varices, ascites
- ~25% have coincidental malformations (polysplenia syndrome: intestinal malrotation, preduodenal portal vein, azygous continuation of IVC)
Diagnosis
No single test is sufficiently sensitive or specific; a combination of studies is used:
- Serum bilirubin fractionation → predominantly conjugated (direct) hyperbilirubinemia
- Liver function tests: elevated GGT, alkaline phosphatase, transaminases
- TORCH titers / viral hepatitis serologies (to exclude infectious causes)
- Abdominal ultrasound:
- Absence of gallbladder — highly suggestive
- Gallbladder presence does NOT exclude BA (present in ~10%)
- No dilated intrahepatic ducts (helps distinguish from choledochal cyst)
- Hepatobiliary scintigraphy (Tc-99m DISIDA/HIDA scan) after phenobarbital pretreatment:
- Radionuclide in intestine = biliary patency → BA excluded
- No excretion into intestine → consistent with BA
- Liver biopsy: portal fibrosis, bile duct proliferation, bile plugs in ducts
- Intraoperative cholangiogram — definitive; confirms obstruction pattern before Kasai procedure
Surgical Treatment — The Kasai Procedure
The hepatoportoenterostomy (Kasai procedure) is the primary treatment:
- The fibrous remnant of the extrahepatic biliary tree is excised at the level of the porta hepatis
- A Roux-en-Y loop of jejunum is anastomosed directly to the transected porta hepatis to drain bile
- Timing is critical: best outcomes when performed before 60 days of age; outcomes deteriorate significantly after 90 days
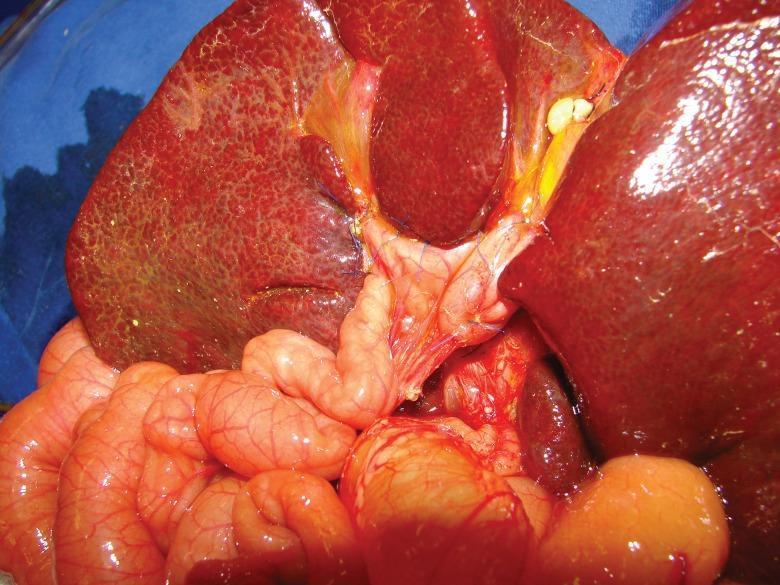
Intraoperative view of a Kasai portoenterostomy: the Roux-en-Y jejunal loop (pink, lower) is sutured to the transected porta hepatis of a cirrhotic liver (dark red/mottled surface).
Post-Kasai outcomes:
- ~50% of patients who undergo timely Kasai will achieve adequate bile drainage
- Even with successful drainage, many develop progressive intrahepatic disease
- Ascending cholangitis is a major post-operative complication (treated with antibiotics; some centers use prophylactic antibiotics)
- Overall: ~30–50% survive to age 10 without liver transplantation after successful Kasai
— Schwartz's Principles of Surgery; Mulholland & Greenfield's Surgery
Liver Transplantation
- Remains the definitive treatment for failed Kasai or end-stage liver disease
- Biliary atresia is the leading indication for pediatric liver transplantation (~50% of all pediatric transplants)
- 5- and 10-year post-transplant survival rates are excellent (>80%)
Key Points Summary
| Feature | Detail |
|---|---|
| Definition | Fibroproliferative obliteration of extrahepatic bile ducts |
| Incidence | 1:8,000–18,000 live births |
| Classic sign | Acholic stools + conjugated jaundice in neonate |
| No dilated intrahepatic ducts | Critical differentiating feature |
| Definitive diagnosis | Intraoperative cholangiogram |
| Treatment | Kasai portoenterostomy (ideally <60 days of age) |
| Definitive cure | Liver transplantation |
| Most common indication for pediatric OLT | Yes (~50–60%) |
— Robbins & Kumar Basic Pathology; Sleisenger & Fordtran's GI and Liver Disease; Schwartz's Principles of Surgery, 11th ed.
This is a shared conversation. Sign in to Orris to start your own chat.