Hodgkin's lymphoma, its classification and its difference and main clinical features and important points for viva and exam point of view
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a comprehensive, exam-oriented guide to Hodgkin Lymphoma (HL):
Hodgkin Lymphoma — Complete Guide
1. Definition & Cell of Origin
Hodgkin Lymphoma (HL) is a B-cell lymphoid neoplasm characterized by the presence of neoplastic Reed-Sternberg (RS) cells in a background of reactive inflammatory cells (lymphocytes, eosinophils, plasma cells, macrophages). Neoplastic RS cells typically make up only <10% of the tumor mass — the majority is reactive infiltrate.
Cell of origin: Germinal center or post-germinal center B cells (proven by microdissection showing clonal IgG gene rearrangements with somatic hypermutation in every RS cell).
2. WHO Classification (5 Subtypes)
| Subtype | Frequency | Key Features |
|---|---|---|
| Nodular Sclerosis (NS) | ~60% | Most common; collagen bands; lacunar RS cells; young females; mediastinum |
| Mixed Cellularity (MC) | ~9% | Classic RS cells; EBV-associated in up to 70%; older males; advanced stage |
| Lymphocyte Rich | ~3% | Best prognosis; background of lymphocytes; few RS cells |
| Lymphocyte Depleted | ~1% | Worst prognosis; many RS cells, few lymphocytes; elderly/HIV |
| Nodular Lymphocyte Predominant (NLPHL) | ~8% | Distinct entity; L&H/"popcorn cells"; CD20+, CD15−, CD30− |
The first four are grouped as Classic Hodgkin Lymphoma (cHL).
— Goldman-Cecil Medicine, Table 172-1; Robbins & Kumar Basic Pathology
3. Reed-Sternberg Cell — The Hallmark
- Classic RS cell: Large binucleated (or multi-nucleated) cell with prominent "owl-eye" nucleoli (large, eosinophilic, inclusion-like)
- Lacunar cell: RS variant in nodular sclerosis; artifact of formalin fixation causes cytoplasmic retraction into a "lacuna"
- L&H (lymphocytic and histiocytic) cell / "Popcorn cell": Seen in NLPHL — multilobated nucleus resembling popcorn
Immunophenotype of RS cells:
| Marker | Classic HL | NLPHL |
|---|---|---|
| CD30 | ✅ Positive (90–100%) | ❌ Negative |
| CD15 | ✅ Positive (75–85%) | ❌ Negative |
| PAX5/BSAP | ✅ Positive (>90%) | ✅ Positive |
| CD20 | ± (~40%, weak/focal) | ✅ Strongly positive |
| CD45 (LCA) | ❌ Negative | ✅ Positive |
| CD79a | ❌ Negative | ✅ Positive |
— Goldman-Cecil Medicine
4. Clinical Features
Presentation
- Painless lymphadenopathy — most commonly cervical (60–70%), then mediastinal, axillary
- Mediastinal mass — especially in nodular sclerosis (young women); can cause SVC syndrome
- B symptoms (present in ~40%):
- Fever >38°C (Pel-Ebstein fever — cyclical, pathognomonic)
- Drenching night sweats
- Weight loss >10% in 6 months
- Pruritus — generalized, may precede diagnosis
- Alcohol-induced pain at lymph node sites — classic but uncommon (pathognomonic when present)
Epidemiology
- Bimodal age distribution: Peak 1 at 25–30 years (young adults); Peak 2 at >50 years
- Slightly more common in males; more common in Whites than Blacks
- Higher incidence in higher socioeconomic classes (Western world)
- In the Indian subcontinent, peak is strongly shifted into childhood
5. HL vs Non-Hodgkin Lymphoma (NHL) — Key Differences
| Feature | Hodgkin Lymphoma | Non-Hodgkin Lymphoma |
|---|---|---|
| Node involvement | Single axial group (cervical, mediastinal, para-aortic) | Multiple peripheral nodes |
| Spread | Orderly, contiguous | Non-contiguous, unpredictable |
| Mesenteric nodes/Waldeyer's ring | Rarely involved | Commonly involved |
| Extranodal presentation | Rare | Common |
| Neoplastic cell | Reed-Sternberg cell | Varied |
| Cell of origin | Germinal center B cell | B cell, T cell, NK cell |
| Prognosis | Generally better, often curable | Variable |
— Robbins, Cotran & Kumar Pathologic Basis of Disease, Table 13.7
6. Pathogenesis & EBV Association
- EBV is the leading suspect: present in RS cells in ~70% of mixed cellularity and ~50% of all classic HL cases
- History of infectious mononucleosis increases HL risk 3-fold
- EBV genome is monoclonal in RS cells of a given patient (infection precedes transformation)
- Immune escape mechanisms:
- Loss of β2-microglobulin → failure to express class I MHC
- High expression of PD-L1 → inhibits T-cell responses (therapeutic target)
- RS cells secrete cytokines: IL-5 (eosinophil attraction), TGF-β (fibrosis), IL-13 (autocrine growth)
7. Staging — Ann Arbor / Lugano Classification
| Stage | Definition |
|---|---|
| I | Single lymph node region or single extralymphatic site |
| II | ≥2 node regions on same side of diaphragm |
| III | Node regions on both sides of diaphragm |
| IV | Disseminated extranodal involvement (bone marrow, liver, lung) |
Modifiers:
- A = No B symptoms; B = B symptoms present
- E = Extranodal contiguous extension; S = Spleen involved
- X = Bulky disease (mediastinal mass >1/3 thoracic diameter or >10 cm)
8. Histopathology of Each Subtype (Exam Focus)
Nodular Sclerosis (most common — 60%)
- Collagen bands divide lymph node into nodules
- Lacunar cells (RS variants) in nodules
- Background: lymphocytes, eosinophils, plasma cells
- Young women, mediastinal involvement
Mixed Cellularity (~9%)
- Classic RS cells scattered among mixed inflammatory background
- Most strongly associated with EBV (up to 70%)
- Older males, advanced stage at presentation
Lymphocyte Rich (~3%)
- Abundant reactive lymphocytes, rare RS cells
- Best prognosis among classic HL
- Resembles NLPHL but RS cells are CD30+, CD15+
Lymphocyte Depleted (~1%)
- Numerous RS cells, depleted lymphocytes
- Worst prognosis; elderly, HIV+ patients
- High association with EBV and advanced stage
Nodular Lymphocyte Predominant (NLPHL, ~8%)
- Popcorn/L&H cells — large neoplastic cells with multilobated nuclei
- Background: predominantly small B lymphocytes and histiocytes
- CD20+, CD79a+, CD45+; CD30−, CD15− ← key distinction
- Indolent course; risk of transformation to diffuse large B-cell lymphoma
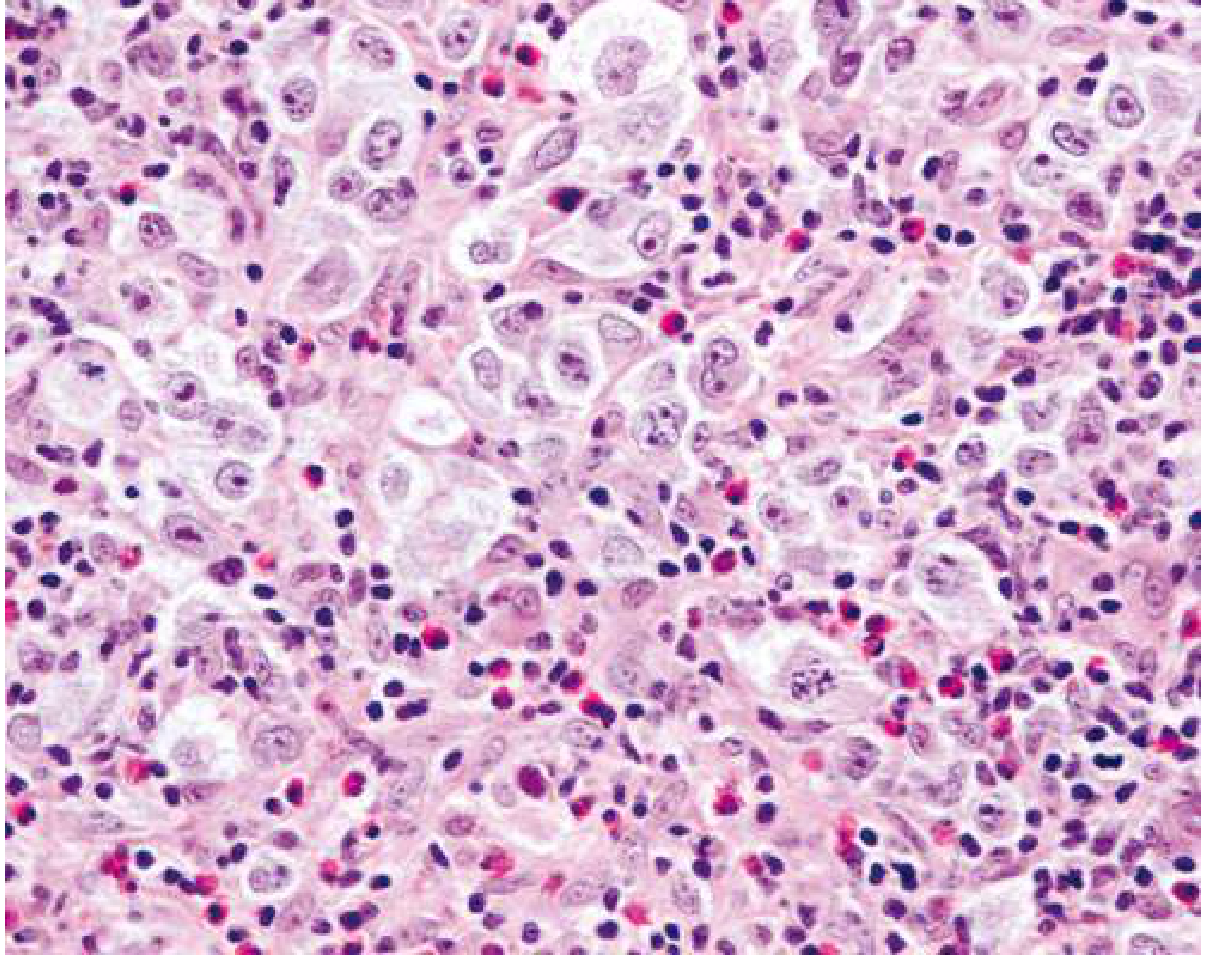
9. Treatment
| Category | Regimen |
|---|---|
| Early-stage (I–II), favorable | 2–4 cycles ABVD + involved-field radiation |
| Early-stage, unfavorable/bulky | 4–6 cycles ABVD ± radiation |
| Advanced stage (III–IV) | 6–8 cycles ABVD; or BEACOPP (escalated, more toxic) |
| Relapsed/refractory | High-dose chemo + autologous stem cell transplant (ASCT) |
| CD30-targeted | Brentuximab vedotin (anti-CD30 antibody-drug conjugate) |
| PD-1 checkpoint | Nivolumab, Pembrolizumab (exploit PD-L1 overexpression on RS cells) |
ABVD = Adriamycin (doxorubicin), Bleomycin, Vinblastine, Dacarbazine
MOPP = historically used; now largely abandoned due to infertility and leukemia risk
MOPP = historically used; now largely abandoned due to infertility and leukemia risk
10. Viva & Exam High-Yield Points
| Point | Detail |
|---|---|
| Most common subtype | Nodular sclerosis (60%) |
| Best prognosis | Lymphocyte rich |
| Worst prognosis | Lymphocyte depleted |
| Most common site of HL | Cervical lymph nodes |
| Classic RS cell | Binucleated, "owl-eye" nucleoli |
| RS cell markers | CD30+, CD15+, PAX5+, CD45− |
| NLPHL markers | CD20+, CD45+, CD30−, CD15− |
| Popcorn/L&H cell | NLPHL |
| Lacunar cell | Nodular sclerosis |
| Bimodal age | 25–30 yrs & >50 yrs |
| Pel-Ebstein fever | Cyclical fever in HL (pathognomonic) |
| Alcohol pain | Pain at nodal sites after alcohol — pathognomonic |
| EBV association | 70% mixed cellularity; ~50% all classic HL |
| HIV association | Lymphocyte depleted subtype; mixed cellularity |
| Cell of origin | Germinal center B cell |
| Spreads | Contiguously (unlike NHL) |
| Mesenteric nodes | Spared in HL; involved in NHL |
| Mediastinal involvement | Nodular sclerosis (young women especially) |
| Transformation risk | NLPHL → DLBCL |
| Treatment backbone | ABVD chemotherapy |
| Second malignancy risk | Alkylating agents → AML/MDS; radiation → solid tumors (breast, lung) |
| Infertility | MOPP/BEACOPP (alkylating agents); ABVD is safer |
| Cardiovascular risk | Mediastinal radiation → coronary artery disease (latency ~19 years) |
Sources: Robbins & Kumar Basic Pathology; Robbins, Cotran & Kumar Pathologic Basis of Disease; Goldman-Cecil Medicine
Mutations
This is a brief follow-up to the Hodgkin Lymphoma topic, so I can answer this from what was already retrieved — the textbooks covered the molecular/mutation aspects in detail.
Mutations in Hodgkin Lymphoma
1. Why RS Cells are Genetically Abnormal B Cells
Normal germinal center B cells that fail somatic hypermutation of their immunoglobulin genes undergo apoptosis. RS cells carry crippling mutations in their rearranged immunoglobulin genes (non-functional V-gene rearrangements) — they should have died but survived due to oncogenic hits that rescue them from apoptosis.
2. Key Signaling Pathways & Mutations
NF-κB Pathway (Most Important)
- Constitutive NF-κB activation is the central oncogenic mechanism in RS cells
- NF-κB promotes RS cell survival, proliferation, and cytokine secretion
- Mechanisms of activation:
- EBV LMP-1 (latent membrane protein-1) directly activates NF-κB in EBV+ cases
- Mutations in TNFAIP3 (A20) — A20 is a negative regulator of NF-κB; loss-of-function mutations in A20 found in ~30–40% of cHL cases
- REL gene amplification — REL encodes a NF-κB subunit; amplified on chromosome 2p
JAK-STAT Pathway
- RS cells show constitutive JAK2 amplification (chromosome 9p24.1)
- JAK2 amplification also co-amplifies PD-L1 and PD-L2 genes on the same locus → immune evasion
- STAT6 activating mutations — found in a subset; STAT6 drives IL-13 autocrine signaling
- SOCS1 mutations — SOCS1 normally inhibits JAK-STAT; loss-of-function mutations lead to sustained STAT activation
PI3K Pathway
- Activated downstream of JAK2 and receptor signaling; contributes to RS cell survival
3. Chromosome 9p24.1 Amplification — High-Yield
This is one of the most exam-important molecular findings in HL:
| Gene at 9p24.1 | Consequence |
|---|---|
| JAK2 | Constitutive kinase activation → proliferation/survival |
| PD-L1 (CD274) | Overexpressed → T-cell exhaustion, immune escape |
| PD-L2 (CD273) | Overexpressed → immune checkpoint activation |
This is why anti-PD-1 agents (nivolumab, pembrolizumab) work so well in relapsed/refractory HL — the tumor is genomically wired to overexpress PD-L1.
4. β2-Microglobulin (B2M) Mutations
- Loss-of-function mutations in B2M → failure to assemble MHC class I molecules
- RS cells become invisible to CD8+ cytotoxic T cells
- A major immune escape mechanism
5. EBV-Driven Oncogenesis (in EBV+ cases)
- LMP-1 mimics a constitutively active CD40 receptor → activates NF-κB and PI3K
- LMP-2A mimics a tonically active B-cell receptor → survival signals without BCR
- Together, LMP-1 and LMP-2A rescue the crippled B cell from apoptosis and drive clonal expansion
6. NLPHL-Specific Mutations
- Unlike classic HL, NLPHL popcorn cells retain functional BCR signaling
- Frequent mutations in TNFRSF14 and RHOA
- Activation of BCR/PI3K pathway — basis for sensitivity to rituximab (anti-CD20)
Summary Table — Viva-Ready
| Mutation / Alteration | Effect | Subtype |
|---|---|---|
| TNFAIP3 (A20) loss | Constitutive NF-κB | Classic HL |
| REL amplification (2p) | NF-κB overactivation | Classic HL |
| JAK2 amplification (9p24.1) | JAK-STAT activation + PD-L1 overexpression | Classic HL |
| STAT6 activating mutation | IL-13 autocrine loop | Classic HL |
| SOCS1 loss | Sustained JAK-STAT | Classic HL |
| B2M loss | MHC-I loss → CTL escape | Classic HL |
| PD-L1/PD-L2 amplification | T-cell checkpoint → immune escape | Classic HL (especially NS) |
| EBV LMP-1 | Constitutive NF-κB + CD40 mimicry | EBV+ HL (MC > NS) |
| EBV LMP-2A | BCR mimicry → survival | EBV+ HL |
| RHOA, TNFRSF14 | BCR pathway activation | NLPHL |
Key viva point: Classic HL survives through two parallel rescue mechanisms — NF-κB activation (prevents apoptosis) and immune evasion via PD-L1 upregulation + MHC-I loss (prevents immune clearance). Both are directly targetable therapeutically.
Numerical and structural mutations in genetics
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I now have comprehensive textbook content on both numerical and structural chromosomal mutations. Here is a complete, well-structured guide:
Numerical & Structural Chromosomal Mutations (Genetics)
PART 1 — NUMERICAL CHROMOSOMAL MUTATIONS
Numerical abnormalities are changes in the total number of chromosomes — either gaining or losing whole chromosomes.
A. Aneuploidy
Definition: Any chromosome number that is NOT an exact multiple of the haploid number (23). The most common and clinically significant numerical chromosomal abnormality — occurs in 3–4% of clinically recognized pregnancies.
| Term | Chromosome Number | Mechanism |
|---|---|---|
| Monosomy | 45 (loss of 1 chromosome) | Nondisjunction |
| Trisomy | 47 (gain of 1 chromosome) | Nondisjunction |
| Nullisomy | 44 (loss of a pair) | Nondisjunction |
| Tetrasomy | 48 (gain of a pair) | Nondisjunction |
Cause: Nondisjunction — failure of a chromosome pair or two chromatids to separate during meiosis I or meiosis II (or mitosis in early embryo).
— The Developing Human: Clinically Oriented Embryology
B. Polyploidy
Definition: Chromosome number is a multiple of 23, but more than 46.
| Type | Number | Cause | Outcome |
|---|---|---|---|
| Triploidy | 69 (3n) | Fertilization by 2 sperm (dispermy) or failure of one meiotic division | Usually lethal; spontaneous abortion |
| Tetraploidy | 92 (4n) | Failure of first cleavage division of zygote | Lethal; blighted embryo (empty chorionic sac) |
C. Mechanism — Nondisjunction
Normal Meiosis: Pair → one to each daughter cell
Nondisjunction: Both go to ONE cell → other gets NONE
Result: One gamete = n+1 (disomic)
Other gamete = n-1 (nullisomic)
Fertilization with normal gamete (n):
n+1 + n = 47 (trisomy)
n-1 + n = 45 (monosomy)
- Can occur in Meiosis I (more common) or Meiosis II
- Can also occur during early mitotic divisions of the zygote → Mosaicism (two or more cell lines with different karyotypes)
D. Clinical Examples of Aneuploidy
Autosomal Trisomies
| Syndrome | Karyotype | Key Features |
|---|---|---|
| Down syndrome | 47, +21 (Trisomy 21) | Most common; upslanting palpebral fissures, epicanthal folds, flat nasal bridge, intellectual disability, Brushfield spots, congenital heart defect (AVSD), single palmar crease |
| Edwards syndrome | 47, +18 (Trisomy 18) | Micrognathia, rocker-bottom feet, clenched fists (overlapping fingers), VSD; 90% die within 1 year |
| Patau syndrome | 47, +13 (Trisomy 13) | Holoprosencephaly, cleft lip/palate, polydactyly, microphthalmia; lethal |
Sex Chromosome Aneuploidies
| Syndrome | Karyotype | Features |
|---|---|---|
| Turner syndrome | 45, X (monosomy X) | Short stature, webbed neck, shield chest, primary amenorrhea, coarctation of aorta, horseshoe kidney, streak gonads |
| Klinefelter syndrome | 47, XXY | Tall, gynecomastia, small testes, infertility, ↓testosterone, learning difficulties |
| Triple X ("Superfemale") | 47, XXX | Usually phenotypically normal female; mild cognitive issues; tall |
| XYY syndrome | 47, XYY | Tall male; usually normal phenotype; behavioral issues reported |
E. Mosaicism
- Nondisjunction during early mitotic divisions post-fertilization
- Results in two cell populations with different karyotypes (e.g., 46,XX / 45,X)
- Phenotype is milder than full monosomy/trisomy
- True hermaphroditism likely due to XX/XY mosaicism
PART 2 — STRUCTURAL CHROMOSOMAL MUTATIONS
Structural abnormalities result from chromosome breakage followed by abnormal reconstitution. Occur in approximately 1 in 375 neonates.
Causes of breaks: Ionizing radiation, viral infections, drugs, chemicals.
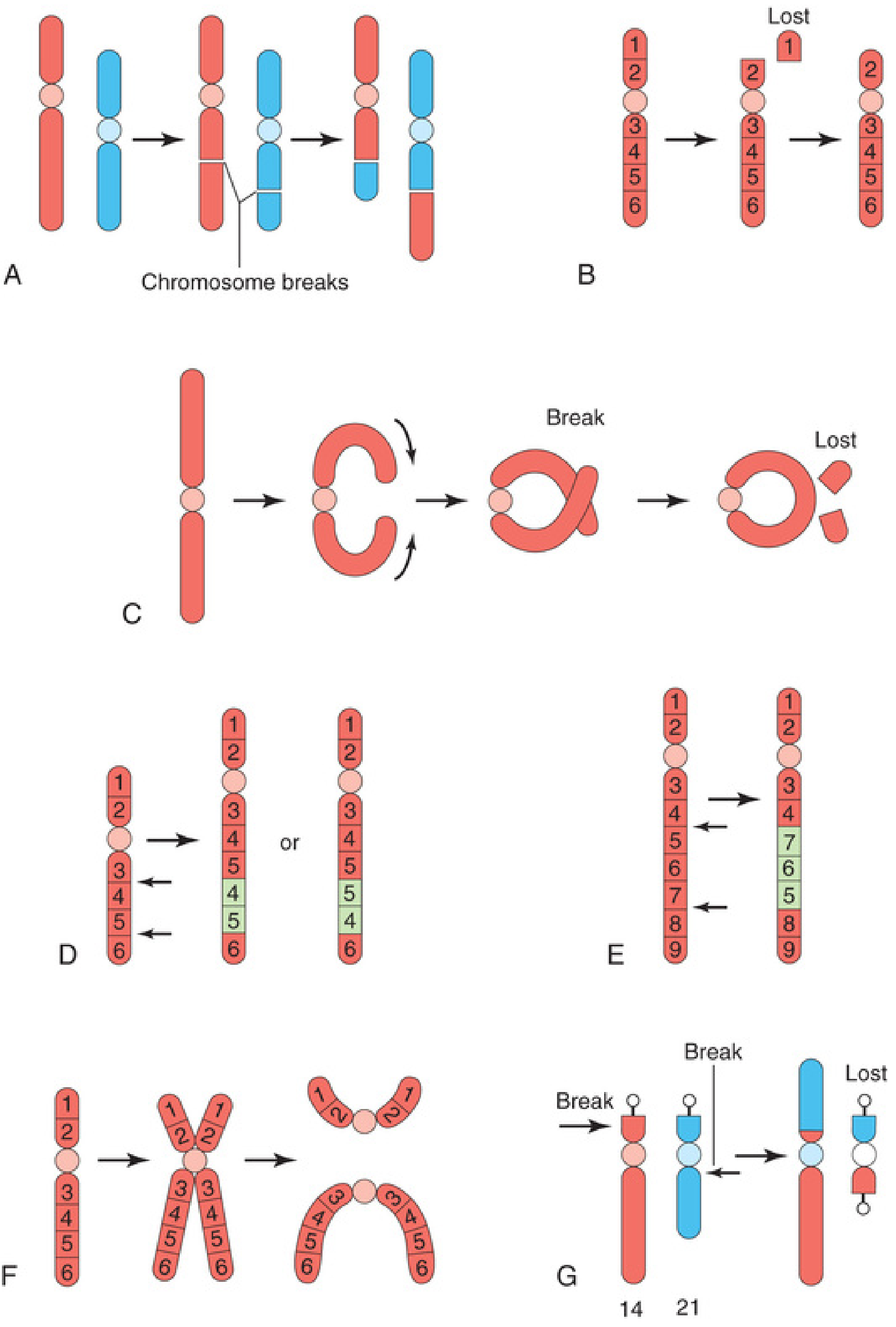
1. Translocation
Transfer of a chromosome segment to a non-homologous chromosome.
| Type | Description | Example |
|---|---|---|
| Reciprocal translocation | Two non-homologous chromosomes exchange pieces | t(9;22) → Philadelphia chromosome in CML |
| Robertsonian translocation | Short arms of two acrocentric chromosomes lost; long arms fuse | t(14;21) → Down syndrome carrier |
- Balanced translocation carrier: Phenotypically normal, but produces abnormal gametes → risk of offspring with unbalanced karyotype
- 3–4% of Down syndrome cases are due to translocation trisomy 21 (not age-related — can recur in family)
- Most common structural abnormality: 1:1000
2. Deletion
Loss of a chromosome segment after breakage.
| Type | Description | Example |
|---|---|---|
| Terminal deletion | Loss from one end of chromosome | Cri du chat syndrome: del(5p) |
| Interstitial deletion | Loss of a middle segment | Di George syndrome: del(22q11) |
| Ring chromosome | Both ends deleted, broken ends join into a ring | Seen in Turner, Edwards syndrome |
Cri du chat syndrome (5p−):
- Cat-like cry (from which the syndrome is named)
- Microcephaly, severe intellectual disability
- Congenital heart disease, hypertelorism
3. Inversion
A chromosome segment is reversed in orientation (requires 2 breaks).
| Type | Description | Clinical Risk |
|---|---|---|
| Paracentric inversion | Confined to ONE arm; does NOT include centromere | Lower risk; acentric/dicentric fragments at meiosis |
| Pericentric inversion | Involves BOTH arms; includes centromere | Higher risk; unequal crossing over → abnormal offspring |
4. Duplication
A chromosome segment is represented twice.
- More common than deletions
- Less harmful than deletions (no genetic material lost)
- Can still cause cognitive impairment or birth defects
- May involve part of a gene, a whole gene, or multiple contiguous genes
5. Isochromosome
A chromosome with two identical arms (both long arms or both short arms) — forms when the centromere divides transversely instead of longitudinally.
- Most common: i(Xq) — isochromosome of long arm of X → one of the causes of Turner syndrome
- Results in monosomy for one arm + trisomy for the other
6. Microdeletion & Microduplication (Contiguous Gene Syndromes)
Very small deletions/duplications detectable only by high-resolution banding or FISH/CMA (chromosomal microarray).
| Syndrome | Chromosome | Deletion/Duplication | Key Features |
|---|---|---|---|
| Prader-Willi | 15q11-q13 | Deletion (paternal) | Obesity, hyperphagia, short stature, hypogonadism, hypotonia |
| Angelman | 15q11-q13 | Deletion (maternal) | Severe intellectual disability, seizures, ataxia, "happy puppet" |
| DiGeorge / velocardiofacial | 22q11.2 | Deletion | Cardiac defects, cleft palate, hypocalcemia, T-cell deficiency |
| Williams | 7q11.23 | Deletion | Elfin facies, supravalvular aortic stenosis, friendly personality |
| Cri du chat | 5p | Deletion | Cat cry, microcephaly, intellectual disability |
| Wolf-Hirschhorn | 4p | Deletion | "Greek warrior helmet" facies, intellectual disability, seizures |
Key concept — Imprinting in 15q11 deletions:
- Same deletion, different parent of origin → different syndrome
- Maternal chromosome 15 deleted → Angelman syndrome
- Paternal chromosome 15 deleted → Prader-Willi syndrome
- This is genomic imprinting — differential expression based on parental origin
SUMMARY TABLE — Viva Quick Reference
| Category | Type | Key Example | Viva Point |
|---|---|---|---|
| Numerical | Trisomy 21 | Down syndrome | Most common cause: nondisjunction in maternal Meiosis I |
| Numerical | Monosomy X | Turner syndrome | Only viable monosomy in humans |
| Numerical | Trisomy 18 | Edwards syndrome | Clenched overlapping fingers |
| Numerical | Trisomy 13 | Patau syndrome | Holoprosencephaly, polydactyly |
| Numerical | XXY | Klinefelter | Infertility, gynecomastia |
| Numerical | Triploidy (69) | — | Dispermy; spontaneous abortion |
| Structural | Reciprocal translocation | t(9;22) CML | Philadelphia chromosome; BCR-ABL |
| Structural | Robertsonian translocation | t(14;21) | Familial Down syndrome; age-independent |
| Structural | Terminal deletion | 5p− | Cri du chat; cat cry |
| Structural | Microdeletion 22q11 | DiGeorge | CATCH-22 |
| Structural | Microdeletion 15q11 (pat) | Prader-Willi | Paternal deletion |
| Structural | Microdeletion 15q11 (mat) | Angelman | Maternal deletion |
| Structural | Pericentric inversion | — | Risk of abnormal offspring |
| Structural | Isochromosome Xq | Turner variant | Transverse centromere division |
| Structural | Ring chromosome | Ring X | Both ends deleted, ends fuse |
Sources: The Developing Human: Clinically Oriented Embryology; Ganong's Review of Medical Physiology; Goldman-Cecil Medicine
This is a shared conversation. Sign in to Orris to start your own chat.