Amyloidosis from pathology point of. view
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Loading Image
Reading File
Loading Image
Reading File
I now have comprehensive, detailed content from Robbins & Kumar. Let me compile this into a thorough pathology-focused answer.
Amyloidosis: A Pathology Perspective
Based primarily on Robbins, Cotran & Kumar - Pathologic Basis of Disease
Definition
Amyloidosis is not a single disease but a group of disorders in which proteins with the propensity to aggregate form insoluble fibrils that deposit in extracellular tissues, causing disruption of tissue architecture and functional compromise. The deposits bind proteoglycans and glycosaminoglycans (heparan sulfate, dermatan sulfate) and plasma proteins, particularly serum amyloid P-component (SAP). The name "amyloid" derives from a historical resemblance to starch (amylose) because of iodine staining - a misnomer since the material is protein, not polysaccharide.
Physical Nature of Amyloid
By electron microscopy, all amyloid - regardless of clinical setting or chemical composition - consists of:
- Continuous, nonbranching fibrils, approximately 8-10 nm in diameter
- Each fibril made up of stacks of protofilaments arranged in a beta-pleated sheet (cross-β) conformation
This beta-pleated sheet configuration is shared by all amyloid types and is directly responsible for the characteristic Congo red staining and birefringence.
Chemical composition: ~95% fibril proteins + ~5% serum amyloid P and other glycoproteins.
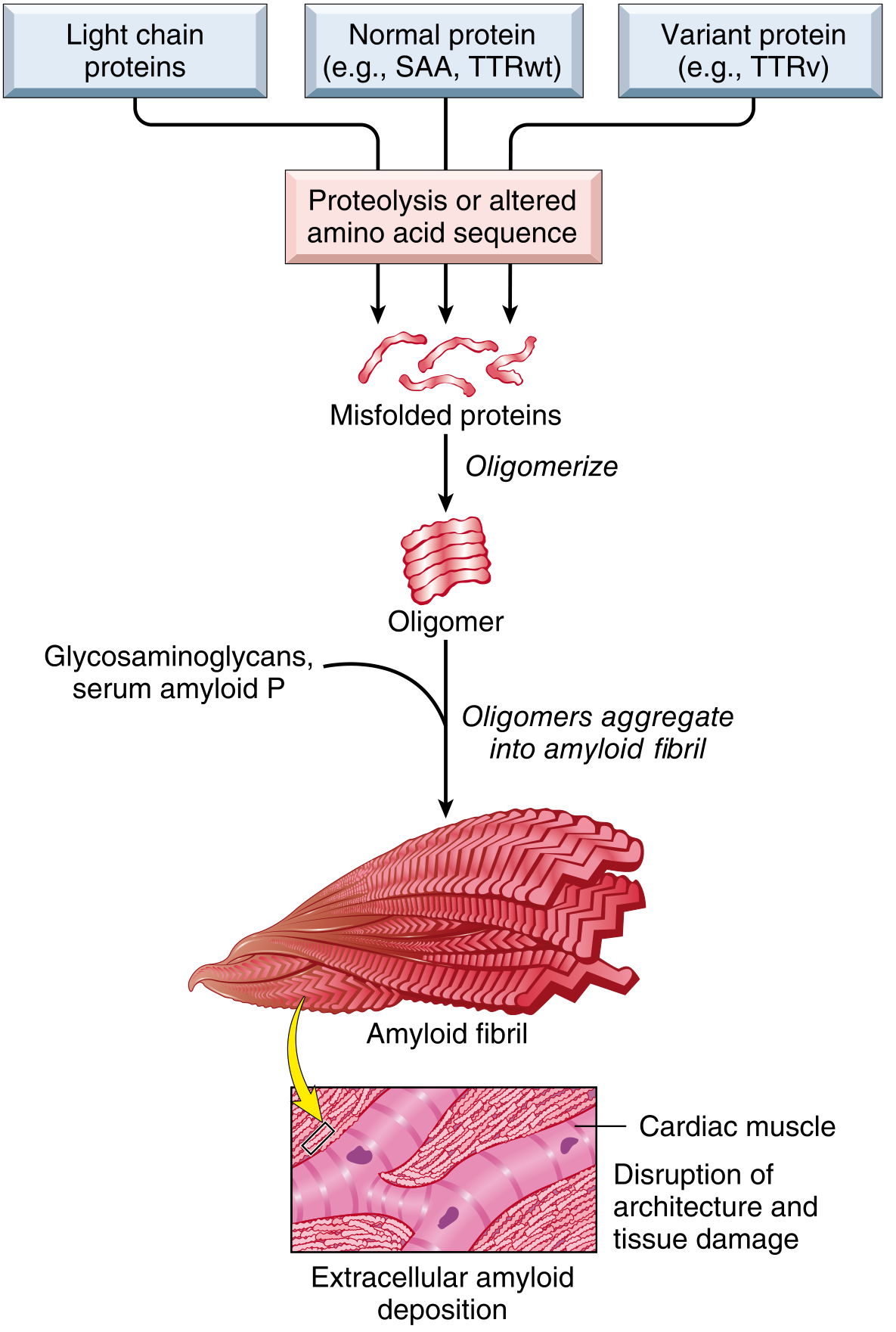
Fig. 6.44 - Mechanism of amyloid deposition (Robbins)
Classification and Pathogenesis
Over 40 different proteins can form amyloid. The three most common forms are:
1. AL Amyloidosis (Amyloid Light Chain)
- Protein source: Monoclonal immunoglobulin light chains (usually lambda > kappa), produced by a clonal plasma cell population
- Clinical setting: Multiple myeloma, other plasma cell dyscrasias, sometimes without overt malignancy (primary amyloidosis)
- Most common systemic form overall
- Preferentially involves: heart, GI tract, respiratory tract, peripheral nerves, skin, tongue
2. AA Amyloidosis (Amyloid-Associated)
- Protein source: Serum amyloid A (SAA), an acute-phase protein synthesized by the liver; proteolytically cleaved to form AA fibrils
- Clinical setting: Chronic inflammatory states - rheumatoid arthritis, inflammatory bowel disease, familial Mediterranean fever, chronic infections (TB, osteomyelitis, bronchiectasis), certain cancers
- Preferentially involves: kidneys, liver, spleen, lymph nodes, adrenals, thyroid
3. ATTR Amyloidosis (Transthyretin)
- Protein source: Transthyretin (TTR), a plasma protein that transports thyroxine and retinol
- ATTRwt (wild-type): Normal TTR aggregates; occurs in males >70 years; cardiac predominance - "senile cardiac amyloidosis"; increasingly prevalent with aging population
- ATTRv (variant/hereditary): Mutant TTR; familial; deposits in heart, peripheral nerves, and other tissues
Other Forms
| Protein | Fibril Name | Setting |
|---|---|---|
| Aβ peptide (from APP) | Aβ | Alzheimer disease (cerebral plaques + vessel walls) |
| β2-microglobulin (MHC I component) | Aβ2m | Long-term hemodialysis - carpal tunnel, joints |
| Misfolded prion proteins | Prion amyloid | Prion diseases (CNS) |
| Calcitonin precursor | ACal | Medullary thyroid carcinoma (local) |
| Insulin | A-insulin | Type 2 diabetes (islets of Langerhans) |
Pathogenic mechanism: Amyloidogenic proteins arise by two mechanisms:
- Normal proteins that have an inherent tendency to misfold when produced in excess or when degradation is impaired (SAA in AA; wild-type TTR in ATTRwt)
- Variant (mutant) proteins that are structurally prone to misfolding (mutant TTR in ATTRv)
Quality-control failure - both intracellular (proteasomal) and extracellular (macrophage-mediated degradation) - allows accumulation of misfolded proteins.
Histopathology and Staining
Light Microscopy (H&E)
Amyloid deposits appear as amorphous, eosinophilic, hyaline, extracellular material - identical in appearance to collagen or fibrin on routine staining alone.
Congo Red Stain (Diagnostic Gold Standard)
- Under ordinary light: Pink-red deposits
- Under polarized light: Characteristic apple-green birefringence
- This reaction is shared by all forms of amyloid and is conferred by the cross-beta pleated fibril configuration
Other Stains
- Crystal violet / methyl violet: Metachromatic staining (purple-red color)
- Thioflavin T/S: Fluorescent staining - bright yellow-green fluorescence under UV (very sensitive, used for frozen sections)
- PAS: Weakly positive, diastase-resistant
- Electron microscopy: Amorphous nonoriented thin fibrils
Specific Typing
- Immunohistochemistry (IHC): Specific antibodies against AA, AL (kappa/lambda light chains), ATTR - required for subtyping
- Mass spectrometry (laser capture + LC-MS/MS): Emerging gold standard for typing, especially when IHC fails
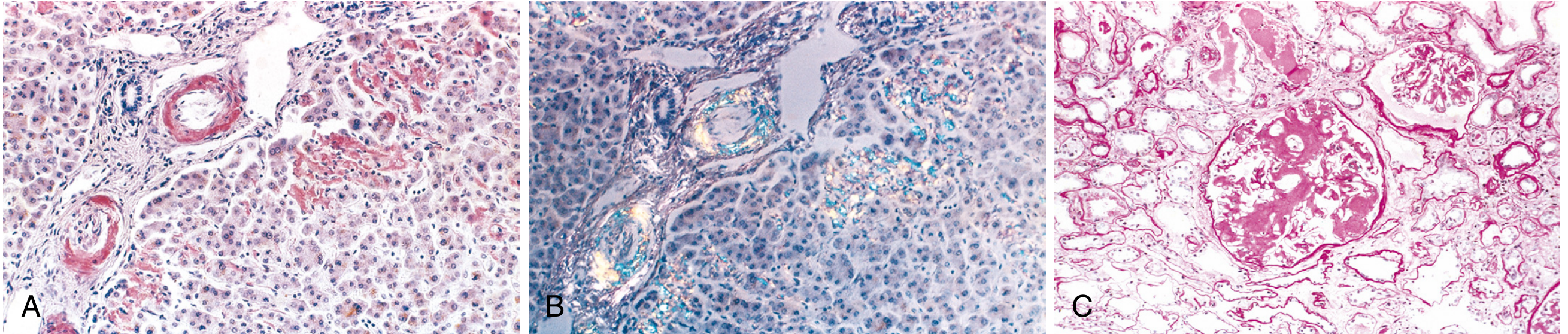
Fig. 6.46 - Amyloidosis (A) Congo red stain in liver, (B) apple-green birefringence under polarized light, (C) massive amyloid obliteration of glomerulus (Robbins)
Morphology by Organ
Gross Appearance
When substantial: enlarged organ, gray, waxy, firm consistency ("lardaceous" organs - from Latin for lard).
Kidney (most common + most serious)
- Early: Subtle mesangial matrix thickening + uneven widening of glomerular basement membranes
- Progressive: Capillary narrowing and distortion of the glomerular vascular tuft
- Advanced: Capillary lumens obliterated; glomerulus flooded by confluent masses of amyloid - "obsolescent glomerulus"
- Interstitium, arteries, and arterioles also involved
- Grossly: normal size or shrunken (from ischemia due to vascular amyloid deposition)
Spleen
Two classic patterns:
- Sago spleen: Deposits limited to splenic follicles; tapioca-like granules on gross inspection
- Lardaceous spleen: Deposits in sinus walls and red pulp connective tissue; large maplike areas; marked splenomegaly (up to 800 g)
Liver
- May be inapparent to massively enlarged
- Deposits begin in space of Disse (between sinusoidal endothelium and hepatocytes)
- Progress to compress hepatic parenchyma
- Portal tracts and hepatic vessels also affected in advanced disease
Heart
- May be enlarged and firm, or no gross change
- Subendocardial focal deposits initially, then between myocardial fibers
- Expansion causes pressure atrophy of myofibers
- Subendocardial deposits damage the conduction system - arrhythmias, heart block
- Produces restrictive cardiomyopathy pattern
Tongue and GI Tract
- Macroglossia: Nodular deposits causing tongue enlargement; characteristic of AL amyloid
- Stomach/intestine: malabsorption, diarrhea
- Vascular fragility: spontaneous or traumatic bleeding; factor X inactivation by AL amyloid causes coagulopathy
Nervous System
- Peripheral and autonomic neuropathies (hereditary ATTR amyloidosis)
- Carpal tunnel syndrome (bilateral) - classic feature of β2-microglobulin amyloidosis (hemodialysis patients)
- Alzheimer disease: cerebral plaques (senile plaques) + cerebrovascular amyloid angiopathy
Adrenals, Thyroid, Pituitary
- Involved in generalized secondary (AA) amyloidosis
Clinical Features
The clinical manifestations depend on the magnitude of deposits and organs affected. Onset is insidious.
| System | Clinical Manifestations |
|---|---|
| Renal | Proteinuria → nephrotic syndrome → progressive renal failure → uremia (common cause of death) |
| Cardiac | Restrictive cardiomyopathy, congestive heart failure, arrhythmias/heart block (can be fatal) |
| GI | Macroglossia, malabsorption, diarrhea, GI bleeding |
| Vascular | Vascular fragility, bleeding; factor X deficiency (AL) → life-threatening coagulopathy |
| Neurologic | Peripheral neuropathy, autonomic neuropathy, carpal tunnel syndrome |
| Constitutional | Weakness, weight loss, lightheadedness, syncope (early and nonspecific) |
Diagnosis
- Biopsy is required for histologic confirmation
- Common biopsy sites:
- Abdominal fat pad aspiration - simple, minimally invasive, ~80% sensitivity in AL/AA
- Rectal biopsy - submucosa sampled
- Kidney - when renal manifestations predominate
- Bone marrow - often positive in AL
- Organ-specific biopsy if above negative but suspicion remains
- SAP scintigraphy (radiolabeled serum amyloid P scan) - whole-body imaging of amyloid burden (not widely available)
- Serum/urine immunofixation + free light chain assay - screening for AL
- Genetic testing - for ATTR variant mutations
Summary Table: Key Amyloid Types
| Type | Fibril Protein | Precursor | Clinical Setting |
|---|---|---|---|
| AL | Light chains (λ > κ) | Immunoglobulins (plasma cells) | Myeloma, plasma cell dyscrasia, primary |
| AA | AA protein | SAA (liver, acute-phase) | Chronic inflammation (RA, IBD, TB, FMF) |
| ATTRwt | Wild-type TTR | TTR | Senile cardiac (males >70) |
| ATTRv | Variant TTR | Mutant TTR gene | Hereditary (FAP, cardiac ATTR) |
| Aβ | β-amyloid | APP (amyloid precursor protein) | Alzheimer disease |
| Aβ2m | β2-microglobulin | MHC-I component | Hemodialysis (carpal tunnel, joints) |
Reference: Robbins, Cotran & Kumar - Pathologic Basis of Disease, Chapter 6 (Diseases of the Immune System), Amyloidosis section, pp. 246-251.
Brain Injuries for Forensic medicine MD exam, SAH,SDH,EDH, concussion, coup, counter coup, differntial diagnosis and short note of Cerbrovascular accident
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Loading Image
Loading Image
Loading Image
Loading Image
I now have all the content needed. Here is the complete forensic medicine MD exam answer:
Brain Injuries - Forensic Medicine MD Exam
Sources: DiMaio's Forensic Pathology 3rd Ed., The Essentials of Forensic Medicine & Toxicology 36th Ed. (2026), Parikh's Textbook of Medical Jurisprudence, Robbins & Kumar Pathologic Basis of Disease, Plum & Posner's Diagnosis and Treatment of Stupor and Coma
1. CONCUSSION
Definition
Concussion is defined as a trauma-induced alteration in mental status that may or may not involve loss of consciousness (Kelley et al.). It is a state of temporary unconsciousness due to head injury, occurring immediately after injury, always followed by amnesia, and tending toward spontaneous recovery.
Mechanism
- Caused by acceleration/deceleration of the head (head must be freely movable at some stage)
- Violent head movement causes shearing or stretching of nerve fibers - diffuse axonal injury
- LOC is caused by transient electrophysiologic dysfunction of the reticular activating system (RAS) in the upper midbrain, produced by rotation of cerebral hemispheres on the relatively fixed brainstem
- At low levels of force: physiological dysfunction only (no anatomical axon damage; may recover)
- At higher force: immediate structural axonal disruption
Grades
| Grade | Features |
|---|---|
| Mild | Confusion, disorientation; NO loss of consciousness; amnesia may/may not occur |
| Severe (Cerebral) | Immediate LOC lasting minutes (usually <6 hours); retrograde + posttraumatic amnesia |
Features During Established Concussion
- Muscles: flaccid
- Pupils: dilated and unreactive
- Pulse: weak and slow
- Respiration: shallow
- On recovery: lucid period where patient appears conscious but acts automatically ("automatic behavior")
- Post-traumatic amnesia (PTA): duration proportional to severity of injury
Postmortem Findings
What is called concussion is thought to be a manifestation of diffuse brain injury with no or insignificant irreparable physical injury. At autopsy, using beta-APP (amyloid precursor protein) staining, multifocal axonal injury can be demonstrated even in "pure" concussion deaths.
Important Forensic Points
- Second impact syndrome: Second head injury before symptoms of first resolve - collapse into coma, ~50% mortality
- Post-concussive apnea: Acute alcohol intoxication + significant head trauma = sudden death; no ICH/skull fracture at autopsy; mechanism = synergistic apnea (BAC 0.168-0.33 g/dL range)
- Concussion can be ruled out if LOC does not occur immediately after blow; delayed coma = other pathology
- "Commotio cerebri": severe head movement causing numerous punctate hemorrhages throughout the brain
2. COUP AND CONTRECOUP INJURIES
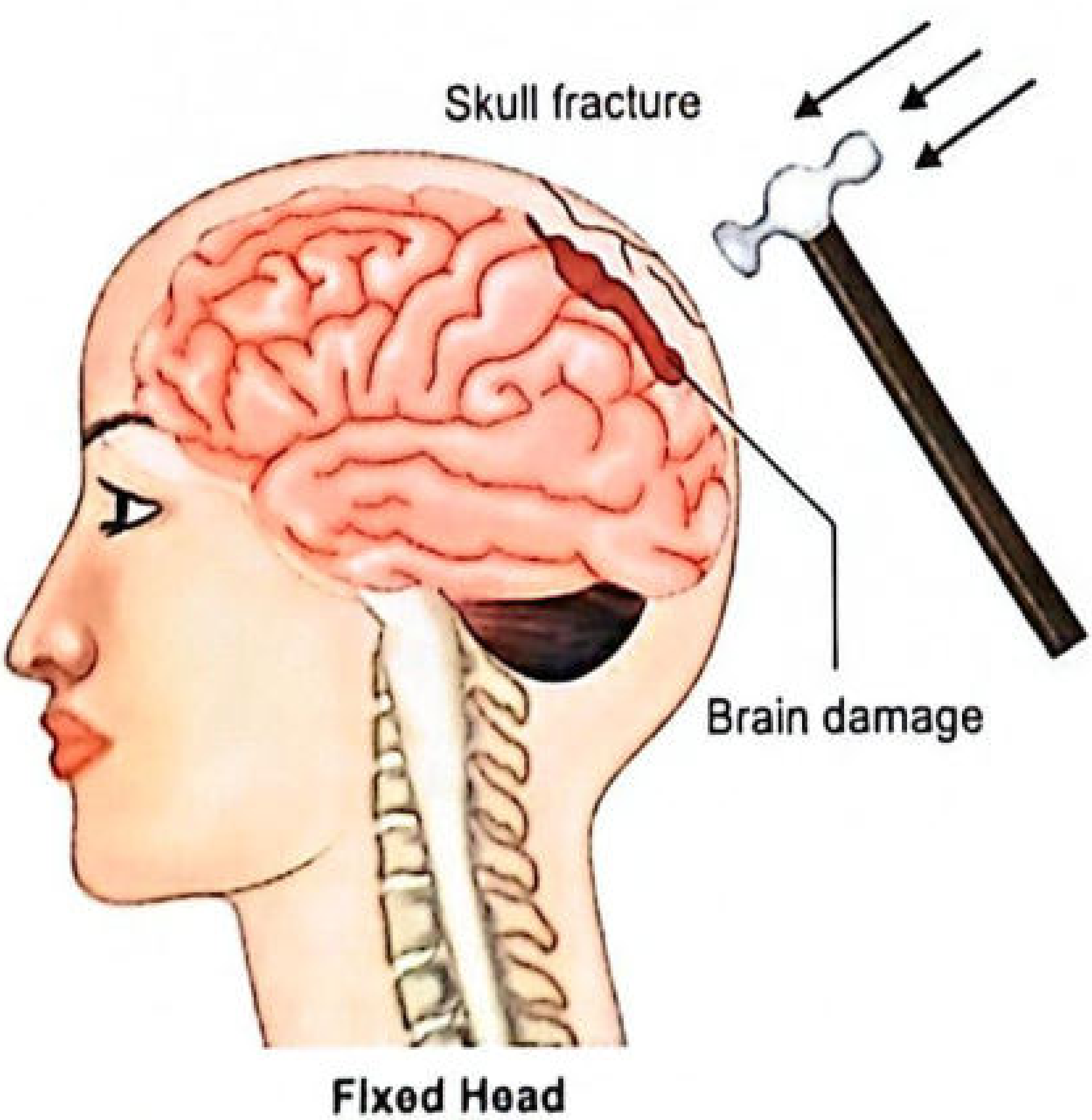
Fig. 9.17 (Essentials of Forensic Medicine & Toxicology): Coup injury mechanism - blow to fixed head
Definitions
- Coup injury: Brain damage located beneath the area of impact - caused directly by the impacting force
- Contrecoup injury (contre = against; coup = blow): Brain damage on the side opposite to the area of impact
Holbourn's Theory (1943)
Contrecoup lesions are chiefly due to:
- Local distortion of the skull and sudden rotation of the head resulting from the blow, causing shear strains (parallel sliding of adjacent tissue layers)
- A much greater shear strain develops as a result of rotation of the skull - since changes in rotational velocity are greater at the pole opposite to impact, contrecoup injuries are more extensive
- Contrecoup can also occur when a blow is struck on a fixed head (e.g., lying on the ground)
Key Forensic Rule
| Mechanism | Coup | Contrecoup |
|---|---|---|
| Blows (moving weapon, fixed head) | Large, pronounced | Small or absent |
| Falls (moving head decelerates) | Small or absent | Large, pronounced |
This allows the forensic pathologist to determine whether injury resulted from a fall or from blows based on the localization of craniocerebral injuries.
Types of Contusion Injuries
- Coup contusions - beneath point of impact
- Contrecoup contusions - opposite side; most common in temporal poles, frontal poles, orbital surfaces of frontal lobes
- Intermediary coup contusions - hemorrhagic contusions in deep brain structures; seen when moving head impacts at the vertex; can mimic spontaneous intracerebral hemorrhage
- Herniation contusions - from brain herniation through tentorial notch
Contrecoup Facts
- Contrecoup can only occur when head is free to move (not pinned against surface)
- Contrecoup injuries are rare before age 3 (due to pliable skull and proportionally larger CSF cushion)
- In temporal impact: contrecoup damage may be on opposite side of ipsilateral hemisphere (against falx cerebri), not the contralateral hemisphere
- There may be no coup damage at all in cases of falls
- Skull fracture may not occur even with severe coup and contrecoup injuries
Gross Morphology of Contusions (Robbins)
- Acute contusions: Hemorrhage and tissue disruption at cortical crests (Robbins Fig. 28.18A)
- Old contusions (Plaque Jaune): Depressed, retracted, yellowish-brown patches on gyral crests - due to hemosiderin accumulation; can become epileptic foci
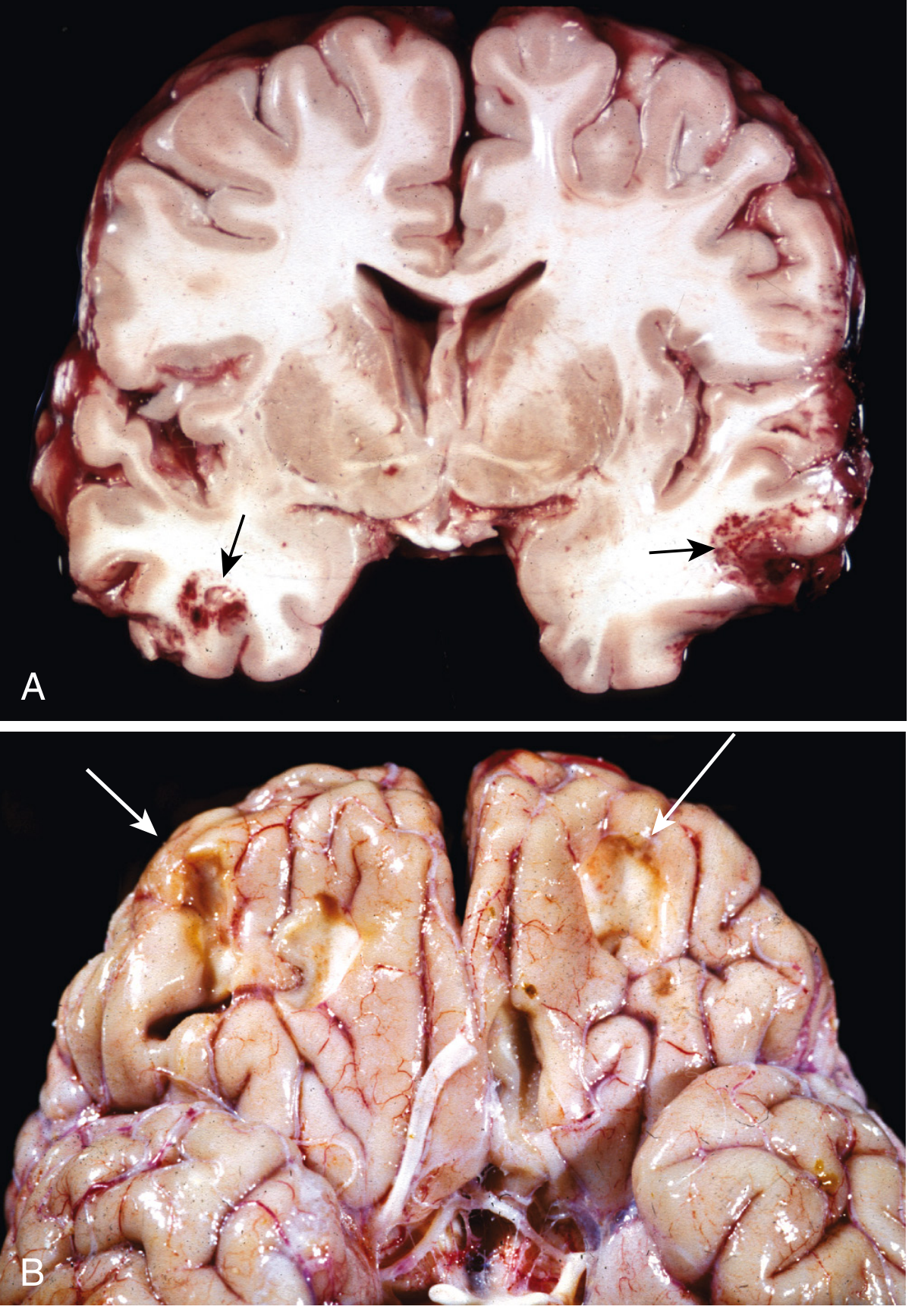
Fig. 28.18 (Robbins): (A) Acute contusions, temporal lobes - hemorrhage and tissue disruption. (B) Remote contusions - plaque jaune (yellowish hemosiderin deposits)
3. EPIDURAL HEMATOMA (EDH)
Definition
Collection of blood between the skull (periosteum) and the outer (periosteal) layer of the dura mater.
Source of Bleeding
- Arterial (most common - 85%): Rupture of the middle meningeal artery following temporal skull fracture (where fracture crosses the arterial groove)
- Venous (15%): From dural venous sinuses or diploic veins; slower development, more self-limited course
Mechanism
- The dura is fused with the periosteum of the inner table of the skull
- Skull fracture (temporal region most common) lacerates the middle meningeal artery
- Arterial blood under high pressure separates dura from periosteum, creating the epidural space
- In children: temporary deformation of the pliable skull can lacerate the vessel without a fracture
Classic Clinical Presentation
"Lucid interval" - Talk and Die"
- Initial LOC or headache at time of injury
- Lucid interval (minutes to hours) - patient awake, orientated
- Sudden neurological deterioration - headache, vomiting
- Ipsilateral pupil dilation (CN III compression by uncal herniation)
- Contralateral hemiplegia
- Coma → death if untreated
Signs of Basal Skull Fracture (associated)
- Battle's sign: Ecchymosis behind the ear (mastoid region) - delayed
- Raccoon eyes: Periorbital ecchymosis
- Blood behind tympanic membrane (hemotympanum)
- CSF rhinorrhea/otorrhea
Morphology
- Biconvex (lenticular) blood collection on CT
- Usually does NOT cross suture lines (dura firmly attached at sutures)
- Most common site: lateral temporal region
4. SUBDURAL HEMATOMA (SDH)
Definition
Collection of blood between the inner layer of dura mater and the arachnoid mater, produced by tearing of bridging veins.
Mechanism
- Bridging veins travel from cerebral convexities through subarachnoid space and dura to the dural sinuses
- The brain is suspended in CSF, but dural sinuses are fixed - traumatic displacement of brain tears the bridging veins where they penetrate the dura
- Extravasated blood dissects through both dural layers
High-Risk Groups
- Elderly (brain atrophy stretches bridging veins) - even minor trauma
- Infants (thin-walled bridging veins) - shaken baby syndrome
- Anticoagulant therapy
- Alcoholics (brain atrophy + coagulopathy)
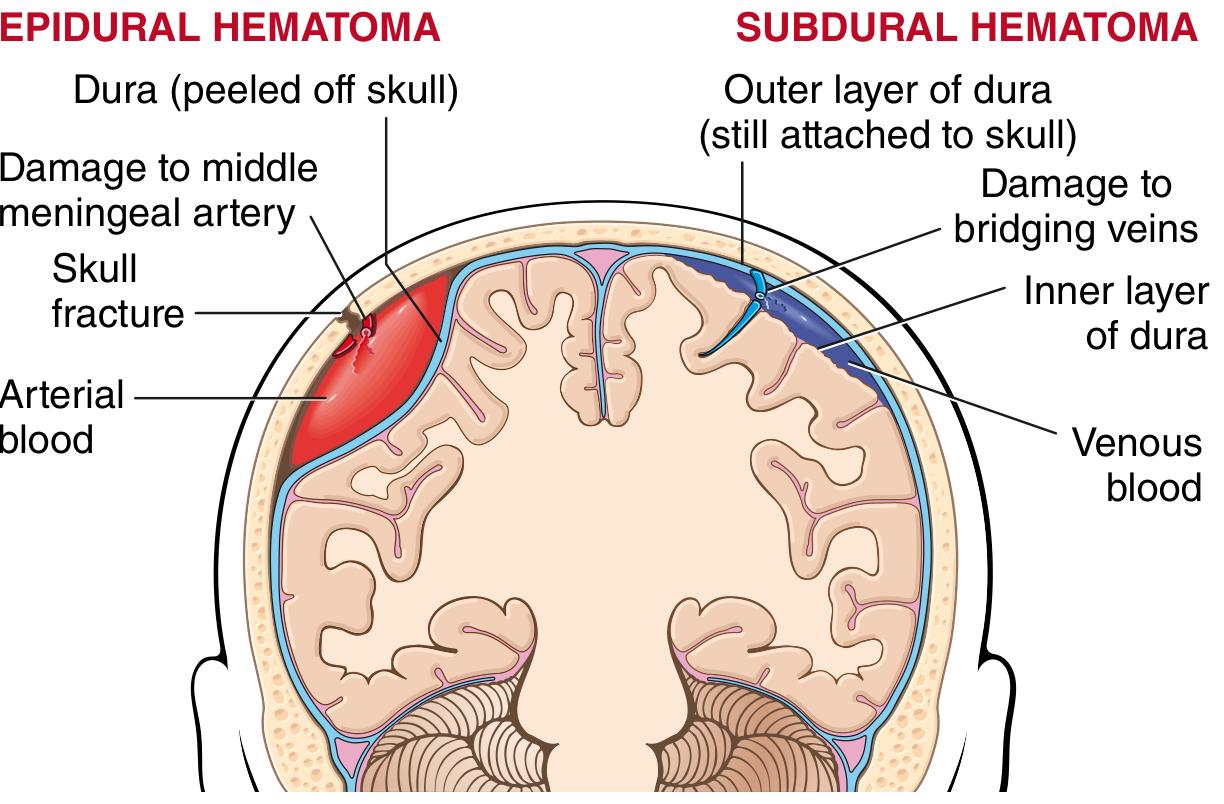
Fig. 28.19 (Robbins): Left - EDH (arterial, biconvex, from middle meningeal artery). Right - SDH (venous, crescent, from bridging veins)
Evolution of Subdural Hematoma (Critical for Forensic Dating)
| Stage | Time | Gross Appearance | Microscopy |
|---|---|---|---|
| Acute | 0-3 days | Freshly clotted blood, red/dark red | Intact RBCs; neutrophil infiltration beginning |
| Subacute | 3 days - 3 weeks | Brownish, liquefying | RBC lysis; hemosiderin; macrophage infiltration |
| Chronic | >3 weeks | Yellow-brown/xanthochromic fluid | Fibrous membranes from dura; neovascularization; no intact RBCs |
Sequence of Organization (Robbins):
- Lysis of the clot (~1 week)
- Fibroblast ingrowth from the dural surface (2 weeks)
- Early hyalinized connective tissue formation (1-3 months)
- "Subdural membranes" - thin reactive connective tissue layer
Chronic SDH: Recurrent bleeding from fragile granulation tissue vessels - can produce multilayered membranes with mixed old and fresh blood. Forensically important: repeated bleeds at different times can indicate ongoing abuse.
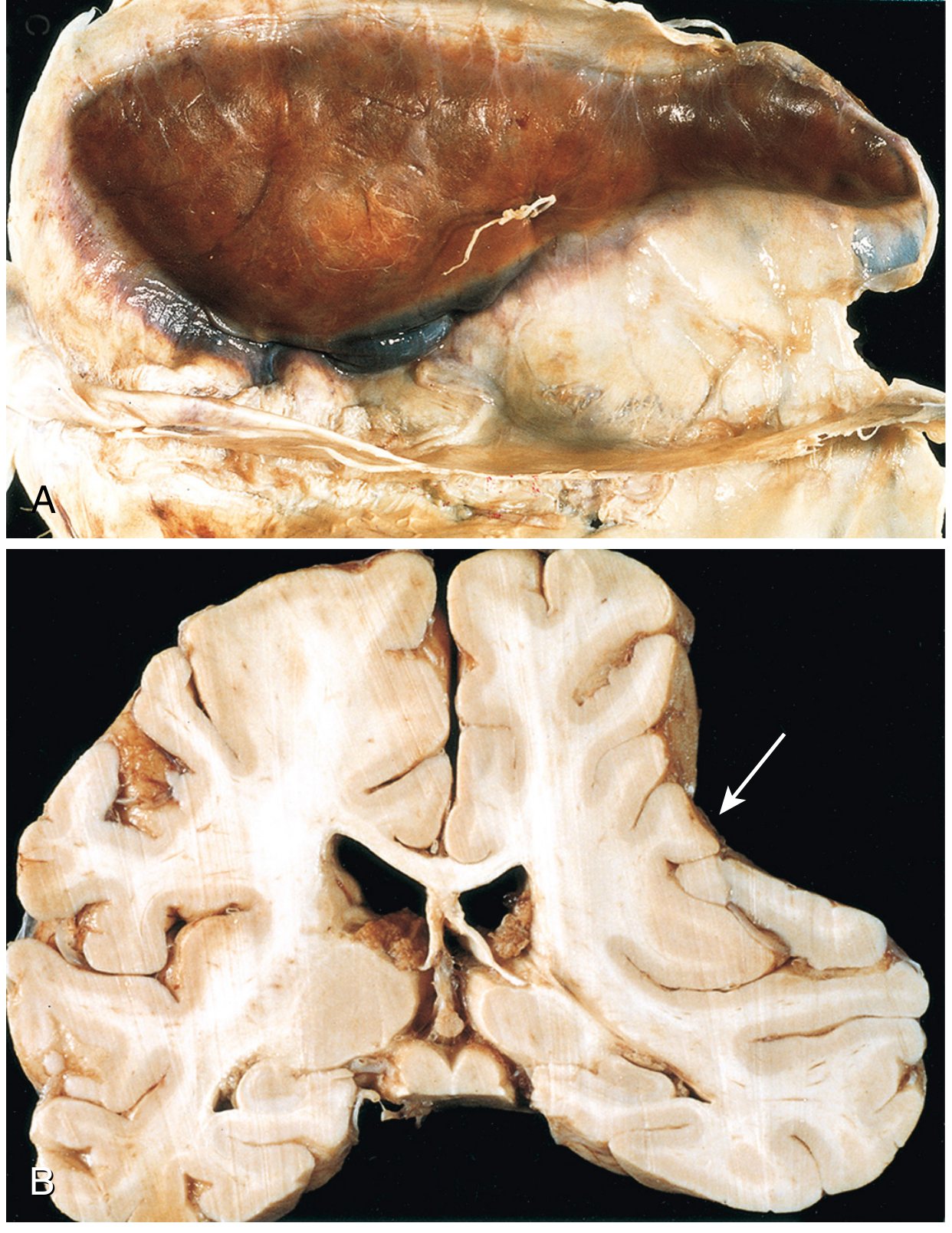
Fig. 28.21 (Robbins): Large organizing SDH - note brain atrophy from compression (B, arrow)
Clinical Presentation
- Symptoms typically within 48 hours of injury
- Slowly progressive neurological deterioration (headache, confusion)
- Nonlocalizing features more common than focal signs
- Bilateral in ~10% of cases
- Acute decompensation can also occur
5. SUBARACHNOID HEMORRHAGE (SAH)
Definition
Bleeding into the subarachnoid space (between the arachnoid mater and pia mater), where CSF circulates.
Causes (Forensic Classification)
| Cause | Examples |
|---|---|
| Traumatic | Most common indicator of TBI; accompanies parenchymal injury; diffuse or focal |
| Natural/Spontaneous | Ruptured berry (saccular) aneurysm (most common), AVM, hypertension |
| Homicidal | Blunt head trauma with SAH as only finding |
| Suicidal | Rare; head injuries |
| Undetermined | Trivial trauma + aneurysm |
Key Forensic Features of Traumatic SAH
- Most common indicator of TBI - can be the only visible sign of brain trauma
- Usually diffuse, overlying the cerebral hemispheres
- In very rapid deaths: tends to be multifocal
- Large collections at the base of the brain (basal cisterns) = more typical of natural disease (ruptured aneurysm) than trauma
- SAH alone can cause death (ruptured berry aneurysm; vertebral artery laceration)
- May cause communicating hydrocephalus - SAH scars the arachnoid villi, impairing CSF reabsorption
Gross Dating of SAH (DiMaio - Forensic Pathology)
| Time Post-injury | Gross Color |
|---|---|
| Acute | Red (fresh blood under leptomeninges) |
| Days | Purplish-black (methemoglobin formation) |
| ~1 week | Brownish (hemoglobin breakdown) |
| Weeks-months | Rust/yellow discoloration (hemosiderin) |
| 6-12 months | Discoloration may disappear (if single SAH event, no associated cortical contusion) |
Microscopic Findings
- Acute: Intact RBCs in subarachnoid space
- Subacute: RBC lysis; xanthochromic CSF (yellow); macrophages with hemosiderin
- Remote: Hemosiderin-laden macrophages; meningeal fibrosis
Forensic Significance
- Traumatic vs. natural SAH - can be very difficult to distinguish at autopsy; extensive blood at base = favor aneurysm; diffuse over convexities = favor trauma
- Trivial trauma + aneurysm - person with undiagnosed berry aneurysm struck a blow; aneurysm ruptures; assailant may face criminal charges - medicolegal nightmare
- "Lucid interval" before death - seen in ruptured aneurysms; may mislead regarding timing
6. DIFFERENTIAL DIAGNOSIS TABLE
EDH vs SDH vs SAH - Key Comparisons
| Feature | EDH | SDH | SAH |
|---|---|---|---|
| Location | Between skull & dura | Between dura & arachnoid | Between arachnoid & pia |
| Source of bleeding | Middle meningeal artery (arterial) | Bridging veins (venous) | Aneurysm/AVM (spontaneous) or pial vessels (traumatic) |
| Shape on CT | Biconvex (lenticular) | Crescent (concave) | Fills basal cisterns / cortical sulci |
| Crosses suture lines | No (dura adherent at sutures) | Yes | N/A |
| Skull fracture | ~85% cases | Not required | Not required |
| Lucid interval | Classic (hours) | Variable (may be absent) | May occur (minutes) |
| Consciousness | Lost → lucid → rapid deterioration | Gradual deterioration | Sudden severe headache ("thunderclap") → deterioration |
| Herniation | Uncal (ipsilateral CN III palsy) | Subfalcine/uncal | Tonsillar / global |
| Age group | Young adults (trauma) | Elderly, infants | 40-60 yrs (aneurysm) |
| Headache quality | Post-trauma headache | Dull, progressive | "Worst headache of life" (thunderclap) |
| Common cause | Fall/assault - temporal blow | Fall in elderly, shaking in infants | Ruptured berry aneurysm |
| Treatment | Urgent surgical drainage | Surgical drainage (acute/large); conservative (chronic small) | Nimodipine, angiography, clipping/coiling |
| Mortality without treatment | Very high (hours) | Variable | 30-50% (aneurysmal) |
Distinguishing Blow vs. Fall (Forensic)
| Finding | Blow (weapon/fist) | Fall (deceleration) |
|---|---|---|
| Coup contusions | Prominent | Small or absent |
| Contrecoup contusions | Small or absent | Prominent |
| Scalp wounds | Localized, often with weapon pattern | Variable |
| Skull fracture | Depressed, localized | Linear, base of skull |
| SAH distribution | Diffuse, over convexities | Diffuse, over convexities |
7. SHORT NOTE: CEREBROVASCULAR ACCIDENT (CVA / STROKE)
Definition
CVA (stroke) is defined as a sudden onset of neurological deficit due to vascular disease of the brain, lasting more than 24 hours (or leading to death), classified as either:
- Ischemic stroke (~80-85%): Infarction due to thrombosis or embolism
- Hemorrhagic stroke (~15-20%): Intracerebral or subarachnoid hemorrhage
Classification
Ischemic Stroke
- Thrombotic: Atherosclerosis of large vessels (carotids, MCA) or small vessel disease (lacunar infarcts)
- Embolic: Cardiac source (AF, mural thrombus post-MI, valvular disease); carotid artery source
- Watershed/hypoperfusion: Systemic hypotension causing infarcts at border zones between major arterial territories
Hemorrhagic Stroke
- Intracerebral hemorrhage (ICH): Hypertension most common (Charcot-Bouchard microaneurysm rupture in basal ganglia, internal capsule, thalamus, pons, cerebellum)
- Subarachnoid hemorrhage (SAH): Berry aneurysm rupture; AVM
Risk Factors
- Hypertension (single greatest risk factor)
- Smoking (doubles lifetime stroke risk; 5-6x risk if hypertensive + smoker)
- Atrial fibrillation, diabetes, dyslipidemia
- OCP (especially with smoking)
- Age, male sex, family history
Morphology
- Hyperacute (0-6h): No gross change; pale infarct beginning; cytotoxic edema
- Acute (12-48h): Pale/swollen cortex, loss of gray-white differentiation; "red neuron" - eosinophilic cytoplasm, pyknotic nucleus
- Subacute (3d - 3wks): Liquefactive necrosis; macrophages (microglial activation); gliosis begins
- Chronic (>1 month): Cavitation/cystic lesion; hemosiderin-laden macrophages; astrocytic gliosis (glial scar)
Forensic Significance
- CVA vs. Head Injury - a person with undiagnosed CVA may fall and sustain head injury; vice versa, head trauma can cause cerebral vasospasm and secondary infarction
- Natural death - intracerebral hemorrhage from hypertension is a natural cause of death; must be distinguished from traumatic hemorrhage
- Position of hemorrhage: Basal ganglia/thalamic/pontine = hypertensive (natural); lobar/subcortical = may be traumatic, amyloid angiopathy, or structural lesion
- Lacunar infarcts at autopsy indicate chronic hypertension - relevant background in establishing pre-existing disease
- Vertebral artery dissection - can occur from trivial trauma (neck manipulation, hyperextension); may cause lethal cerebellar/brainstem infarction or SAH - major medicolegal significance
TIA (Transient Ischemic Attack)
- Neurological deficit resolving within 24 hours (by old definition) or <1 hour with no infarct on imaging (new definition)
- Harbinger of stroke - 10-15% risk of stroke within 3 months of TIA; highest risk in first 48 hours
- Forensically: TIA preceding fatal stroke may be documented in medical records - establishes pre-existing vascular disease
SUMMARY MNEMONIC TABLE
| Injury | Vessel/Source | Space | CT Shape | Classic Sign |
|---|---|---|---|---|
| EDH | Middle meningeal A. | Epidural | Biconvex | Lucid interval; temporal fracture |
| SDH | Bridging veins | Subdural | Crescent | Elderly + minor trauma; evolves over days-weeks |
| SAH | Aneurysm/pial vessels | Subarachnoid | Fills cisterns/sulci | Thunderclap headache; neck stiffness |
| Concussion | No gross hemorrhage | Diffuse axonal | Normal CT | Immediate LOC + retrograde amnesia |
| Coup | Cortical vessels | Cortex at impact | Hemorrhagic contusion | Beneath impact site |
| Contrecoup | Cortical vessels | Cortex opposite | Hemorrhagic contusion | Opposite impact; larger in falls |
References: DiMaio's Forensic Pathology 3rd Ed. (pp. 145-152); The Essentials of Forensic Medicine & Toxicology 36th Ed./2026 (pp. 250-260); Parikh's Medical Jurisprudence (Ch. 20); Robbins & Kumar Pathologic Basis of Disease (pp. 1160-1164, Chapter 28)
write an answer on Pathophysiology of shock using harrison and forensic material with shock kidney short note,
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Reading File
Reading File
Reading File
Pathophysiology of Shock
Sources: Harrison's Principles of Internal Medicine 22nd Ed. (2025) - Ch. 314; Schwartz's Principles of Surgery 11th Ed. - Ch. 5; The Essentials of Forensic Medicine & Toxicology 36th Ed. (2026); P.C. Dikshit's Forensic Medicine & Toxicology
Definition
Shock is the clinical condition of organ dysfunction resulting from an imbalance between cellular oxygen supply and demand, resulting in cellular and tissue hypoxia. (Harrison's, 22E)
Schwartz's defines it more surgically: "the failure to meet the metabolic needs of the cell and the consequences that ensue." As Samuel V. Gross wrote in 1872 - "Shock is the manifestation of the rude unhinging of the machinery of life."
Classification of Shock (Harrison's Table 314-1)
| Type | Primary Derangement | Examples |
|---|---|---|
| Distributive | Reduced SVR; compensatory ↑CO | Septic shock, anaphylaxis, neurogenic, burns, pancreatitis, adrenal crisis |
| Cardiogenic | Pump failure; reduced CO | MI, myocarditis, arrhythmia, severe valve disease |
| Hypovolemic | Reduced preload/circulating volume | Hemorrhage (trauma, GI bleed), GI losses, burns, DKA, diabetes insipidus |
| Obstructive | Mechanical obstruction to flow | Tension pneumothorax, cardiac tamponade, PE, aortic dissection, constrictive pericarditis |
Determinants of Oxygen Delivery (DO₂) - Harrison's Framework
The entire pathophysiology of shock can be understood through the DO₂ equation:
DO₂ = CO × CaO₂
where:
CO = HR × SV
SV α (Preload × Contractility) / SVR
CaO₂ (mL/dL) = (Hb × 1.34 × SaO₂) + (PaO₂ × 0.003)
Any disease process affecting HR, preload, contractility, SVR, SaO₂, or Hb reduces oxygen delivery and can cause cellular hypoxia. Each shock type has a distinct hemodynamic profile based on which variable is primarily disturbed.
Hemodynamic Profiles (Schwartz Table 5-2)
| Type | Cardiac Index | SVR | CVP/PCWP | SvO₂ |
|---|---|---|---|---|
| Hypovolemic | ↓ | ↑ | ↓ | ↓ |
| Cardiogenic | ↓↓ | ↑↑ | ↑ | ↓ |
| Septic (Distributive) | ↑↑ | ↓ | ↑↓ (variable) | ↑↓ |
| Neurogenic | ↑ | ↓ | → | ↓ |
Pathophysiology - Cellular Level
1. Oxygen Debt and Anaerobic Metabolism
With insufficient oxygen supply, the cell cannot sustain aerobic metabolism:
- Normal aerobic pathway: Glucose → Pyruvate → Krebs cycle → Oxidative phosphorylation → 38 ATP per glucose molecule
- Anaerobic shift in shock: Glucose → Pyruvate → Lactate → only 2 ATP per glucose
- The result is intracellular metabolic acidosis, ATP depletion, and lactate accumulation
The concept of oxygen debt (Crowell, 1961): the cumulative deficit in tissue oxygenation over time during shock - the larger and longer the debt, the greater the risk of irreversible injury and death.
2. Failure of ATP-Dependent Mechanisms
Depletion of ATP disrupts:
- Na⁺/K⁺ ATPase pump (consumes 20-80% of cellular ATP) → cellular swelling, loss of membrane potential
- Influx of Ca²⁺ → activation of calcium-dependent phospholipases and proteases → cellular swelling and death
- Intracellular pH falls → altered enzyme activity, impaired ion exchange, disrupted metabolic signaling
- DNA repair mechanisms and protein synthesis fail
- Leakage of intracellular contents into extracellular space activates inflammatory cascades
3. Microcirculatory Dysfunction
- Capillary leak: Increased permeability → intracellular swelling → extracellular fluid deficit
- Dysfunction of Na⁺/K⁺ pump causes loss of membrane integrity and fluid shifts
- Endothelial activation by circulating inflammatory mediators → leukocyte adherence → capillary occlusion ("no-reflow phenomenon")
- No-reflow may persist even after macroscopic restoration of perfusion - ongoing ischemia
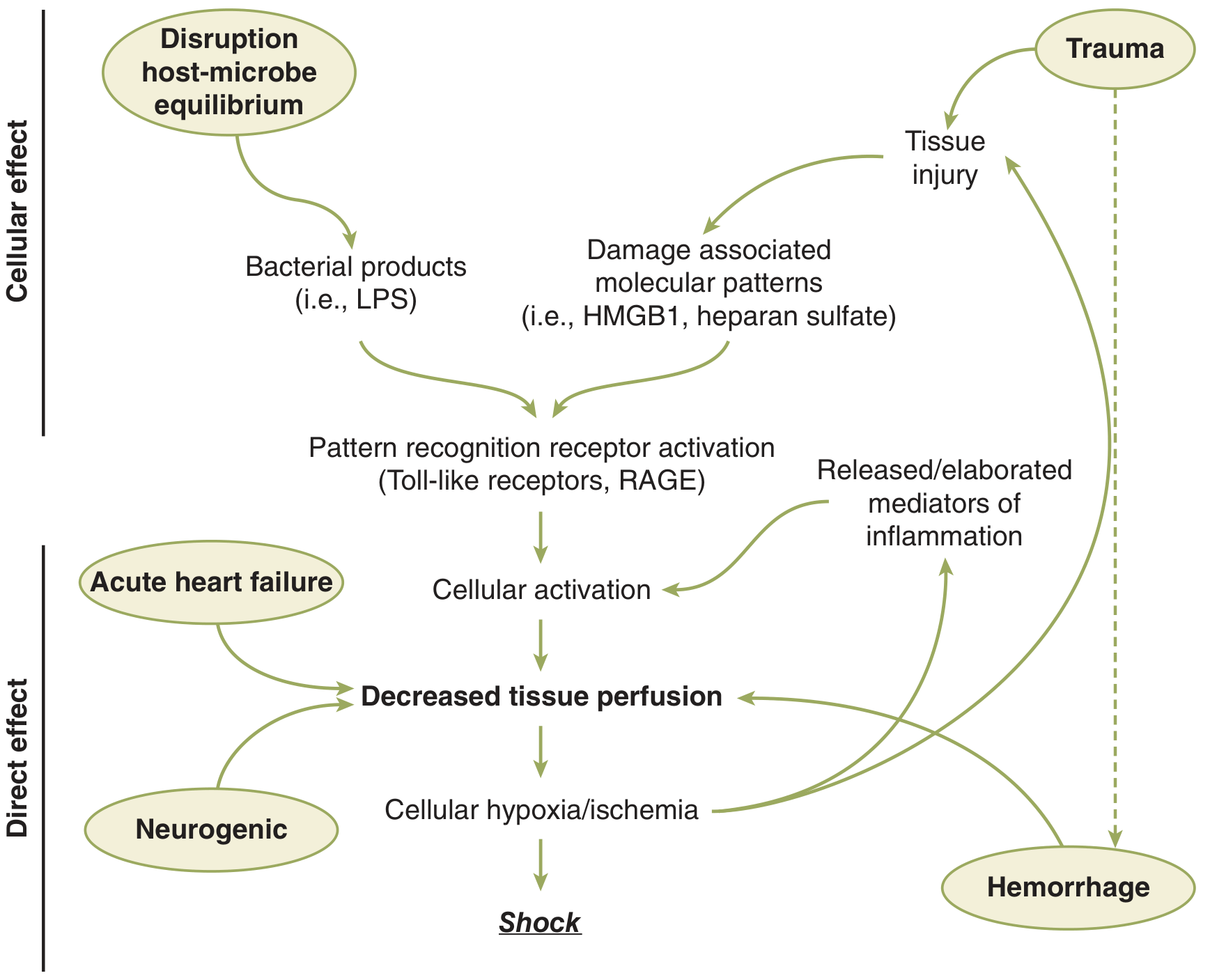
Fig. 5-1 (Schwartz): Pathways leading to decreased tissue perfusion and shock. HMGB1 = high mobility group box 1; LPS = lipopolysaccharide; RAGE = receptor for advanced glycation end products.
Phases of Shock (Schwartz)
Phase 1 - Compensated Shock
- Body compensates for initial blood/volume loss primarily via the neuroendocrine response
- Hemodynamics partially maintained; clinical signs may be subtle
- Reversible with prompt treatment
Phase 2 - Decompensated Shock
- Continued hypoperfusion → cellular death and injury ongoing
- Microcirculatory dysfunction, parenchymal tissue damage, inflammatory cell activation perpetuate hypoperfusion
- Ischemia/reperfusion injury exacerbates the initial insult
- Forms the "vicious cycle of shock" - decreased perfusion → cellular injury → inflammatory response → further perfusion impairment
Phase 3 - Irreversible Shock
- Extensive enough parenchymal and microvascular injury that volume resuscitation fails to reverse the process
- MODS (multiorgan dysfunction syndrome) → death
- In experimental models (modified Wiggers model): occurs when shed blood volume must be continually returned to maintain set hypotension level
Neuroendocrine Response to Shock (Afferent Signals)
The goal of the neuroendocrine response is to maintain perfusion to the heart and brain, even at the expense of other organs.
Afferent Triggers
- Volume/pressure receptors (baroreceptors):
- Low-pressure receptors in cardiac atria - activated by low-volume hemorrhage/mild ↓ right atrial pressure
- High-pressure receptors in aortic arch and carotid sinus - respond to larger ↓ in intravascular volume/pressure
- Normally inhibit ANS; when activated by hypotension → disinhibit ANS → sympathetic activation
- Chemoreceptors (aorta, carotid bodies): Sensitive to ↓O₂ tension, ↑H⁺, ↑CO₂
- Pain signals: Via spinothalamic tracts → hypothalamic-pituitary-adrenal (HPA) axis activation
- DAMPs and PAMPs: Damage-associated molecular patterns (HMGB1, heparan sulfate) and bacterial products (LPS) activate TLRs and RAGE receptors → inflammatory cascades
Efferent Responses
Cardiovascular Response
- ↑ Heart rate and contractility via β₁-adrenergic receptor activation (sympathetic)
- Peripheral vasoconstriction via α₁-adrenergic receptors on arterioles → ↑ SVR
- Blood redistribution: Blood shunted away from intestine, kidney, and skin → preserved to heart and brain (autoregulation)
- Venous constriction → ↓ venous capacitance → ↑ venous return → ↑ preload
Hormonal Response (HPA Axis)
- Hypothalamus → CRH → Pituitary → ACTH → Adrenal cortex → Cortisol
- Cortisol effects: gluconeogenesis, insulin resistance, hyperglycemia, protein breakdown, lipolysis, sodium and water retention
- Catecholamines (epinephrine from adrenal medulla; norepinephrine from sympathetic synapses) peak at 24-48 hours - hepatic glycogenolysis, gluconeogenesis, ↓ insulin, ↑ glucagon → hyperglycemia
- ADH (vasopressin): Released from posterior pituitary → ↑ water retention at collecting duct, vasoconstriction; also ↑ hepatic gluconeogenesis
- RAAS: ↓ renal perfusion → ↑ renin → angiotensin II (vasoconstriction) → aldosterone → ↑ Na⁺ and water retention
Net Metabolic Effect
A catabolic state: glucose mobilization, hyperglycemia, protein breakdown, negative nitrogen balance, lipolysis, and insulin resistance - designed to preserve glucose for heart and brain.
Immune and Inflammatory Responses
Cytokines in Shock
| Proinflammatory | Anti-inflammatory |
|---|---|
| TNF-α, IL-1α/β | IL-4, IL-10, IL-13 |
| IL-2, IL-6, IL-8 | Prostaglandin E₂ |
| Interferon-γ | TGF-β |
| PAF, Thromboxane A₂ | IL-1 receptor antagonist |
| Leukotrienes |
- TNF-α (cachectin): First cytokine released in shock; produced by macrophages; mediates endothelial damage, coagulation activation, fever, hypotension
- IL-1: Short half-life (6 min); paracrine actions; fever (via prostaglandins in hypothalamus); augments ACTH, glucocorticoids, β-endorphins; synergizes with TNF-α
- IL-6: Acute-phase protein synthesis (including SAA, fibrinogen, CRP); prolonged circulation
- NF-κB: Master transcription factor for proinflammatory gene expression, activated by LPS via TLR4/MyD88 pathway
Complement Activation
- C3a and C5a (anaphylatoxins): mast cell degranulation, ↑ vascular permeability, smooth muscle contraction
- C5a: potent neutrophil chemotaxis; upregulates adhesion molecules on endothelium
Neutrophil Activation
- Neutrophils adhere to activated endothelium via ICAM-1/integrin interactions
- Release reactive oxygen species (ROS), proteases, elastase → tissue damage
- Neutrophil depletion in animal models of hemorrhagic shock → fewer no-reflow capillaries and lower mortality
Organ-Specific Responses in Shock
Kidney
- Early: Afferent arteriolar vasoconstriction → ↓ GFR → ↑ renin-angiotensin-aldosterone → Na⁺ and water conservation
- Renal blood flow preferentially shunted away (sympathetic vasoconstriction)
- Persistent ischemia → acute tubular necrosis (see Shock Kidney, below)
Liver
- Normally receives 25% of CO (dual supply: portal vein + hepatic artery)
- In shock: portal flow ↓ dramatically; hepatic artery flow partially maintained
- Ischemic hepatitis ("shock liver"): ↑↑ AST/ALT, ↓ synthesis of clotting factors, albumin
- Kupffer cell activation → release of TNF-α, IL-1, IL-6 → systemic inflammatory amplification
- Lactate clearance is impaired - worsening lactic acidosis (Fig 5-8 Schwartz: progressive ↑ serum, muscle, and liver lactate in hemorrhagic shock)
Lung
- Acute lung injury (ALI) / ARDS: Endothelial injury → diffuse alveolar damage → ↑ permeability pulmonary edema
- Neutrophil sequestration in pulmonary capillaries → oxidant-mediated injury
- Surfactant inactivation by free radicals → atelectasis, refractory hypoxemia
- Common cause of death in patients surviving initial resuscitation from shock
Gastrointestinal Tract
- Gut: "motor" of MODS - highest sensitivity to ischemia
- Intestinal mucosa normally relies on high blood flow (25% of CO at rest)
- Ischemia → disruption of mucosal barrier integrity
- Bacterial/endotoxin translocation from the gut lumen → portal blood → systemic circulation → amplifies SIRS
- Stress ulcers (Curling's ulcers in burns), ileus, mucosal hemorrhage
Adrenals
- Hyperactivated initially (HPA axis); in prolonged severe shock → adrenal insufficiency (relative or absolute)
- Relative adrenal insufficiency in septic shock: major contributor to vasopressor-refractory hypotension
SHORT NOTE: SHOCK KIDNEY (Lower Nephron Nephrosis)
Definition
Shock kidney (also called lower nephron nephrosis or acute tubular necrosis - ATN) is acute renal failure resulting from ischemic damage to renal tubular epithelium, particularly the distal tubules (lower nephron), following prolonged shock of any cause.
The term "lower nephron nephrosis" was coined because early studies noted pigment casts and degenerative changes concentrated in the distal convoluted tubule, loop of Henle, and collecting duct - the "lower nephron."
Causes (Forensic Context)
| Cause | Mechanism |
|---|---|
| Hemorrhagic/hypovolemic shock | Renal ischemia from sustained hypoperfusion |
| Crush syndrome | Myoglobinuria (pigment nephropathy) + ischemia |
| Burns | Plasma loss → hypovolemia + hemolysis + myoglobin |
| Incompatible blood transfusion | Hemoglobinuria + immune complex deposition |
| Septic shock | Ischemia + direct endotoxin tubular toxicity |
| Toxic (Hg, CCl₄, phenol, dextran) | Direct tubular epithelial toxicity + ischemia |
| Obstetric accidents | Placenta previa, abruptio placentae, eclampsia, septic abortion → DIC + ischemia |
Forensically: Lower nephron nephrosis complicating burns typically appears on the 3rd or 4th day post-burn. It is a recognized cause of death in surviving burn patients and a standard finding to document at autopsy in those dying of delayed burns/shock.
Pathogenesis
- Renal vasoconstriction: Sympathetic activation + angiotensin II + endothelin → ↓ renal blood flow → ischemia of tubular epithelium
- Tubular cell injury: ATP depletion → cellular swelling → loss of brush border → loss of tight junctions → "back leak" of filtrate
- Tubular obstruction: Sloughed epithelial cells + protein casts (myoglobin, hemoglobin) block tubular lumen → ↑ intratubular pressure → further ↓ GFR
- Afferent arteriolar vasoconstriction (macula densa feedback): Back-leaked filtrate detected by macula densa → tubuloglomerular feedback → further ↓ GFR
- DIC: Often accompanies severe shock → fibrin thrombi in glomerular capillaries → cortical necrosis in severe cases
Morphology
| Feature | Finding |
|---|---|
| Gross | Kidneys swollen, pale cortex, congested dark medulla; cortex bulges on section |
| Histology - Cortex | Glomeruli relatively normal in early ATN; tubular epithelial swelling, vacuolation |
| Histology - Medulla | Degenerative changes in distal tubules: loss of brush border, nuclear pyknosis, "ghost cells," coagulative necrosis |
| Tubular casts | Pigment casts (myoglobin/hemoglobin), granular casts, epithelial cell casts in distal tubules and collecting ducts |
| Interstitium | Edema, mild inflammatory cell infiltrate |
| Patchy tubular necrosis | Patchy distribution - proximal + distal segments affected; sparing of some adjacent tubules |
Clinical Stages (Forensic Dating of Renal Failure Post-Shock)
| Stage | Duration | Features |
|---|---|---|
| Oliguric phase | 1-3 weeks (avg. ~10 days) | Urine output <400 mL/day; ↑ BUN, creatinine; hyperkalemia, acidosis; fluid overload |
| Diuretic phase | 1-2 weeks | ↑ urine output (may be 3-5 L/day); tubules recovering but concentrating ability lost; risk of hypokalemia, dehydration |
| Recovery phase | Weeks to months | Progressive return of concentrating ability; most patients recover completely if they survive |
Forensic Significance
- Autopsy diagnosis of shock kidney indicates the deceased survived the initial insult long enough to develop ATN (minimum 12-24 hours survival post-insult)
- Tubular necrosis + pigment casts in a burn victim or crush injury victim at autopsy = death from complications of shock
- Cortical necrosis (bilateral) = irreversible injury; seen in severe obstetric DIC, prolonged surgical shock - indicates the kidney received no perfusion for an extended period
- Dextran toxicity (a recognized forensic finding): Intravenous dextran used as plasma expander can itself cause lower nephron nephrosis - medically significant
- Mercury/mercuric chloride poisoning can produce a picture similar to (or identical to) lower nephron nephrosis - important differential at autopsy in suspected poisoning
Summary Diagram: Cascade of Shock Pathophysiology
PRECIPITATING CAUSE
(Hemorrhage / Infection / Cardiac failure / Obstruction)
↓
↓ Cardiac Output and/or ↓ SVR
↓
↓ Oxygen Delivery to Tissues (DO₂)
↓
CELLULAR HYPOXIA
• Aerobic → Anaerobic metabolism
• ATP depletion
• Lactic acidosis
• Membrane pump failure
• Ca²⁺ influx → cell death
↓
NEUROENDOCRINE RESPONSE INFLAMMATORY RESPONSE
• ↑ SNS → ↑ HR, ↑ SVR • TNF-α, IL-1, IL-6
• ↑ Catecholamines • Complement activation
• ↑ Cortisol, ADH • Neutrophil sequestration
• ↑ RAAS → Na⁺/H₂O retention • NF-κB activation
↓
ORGAN-SPECIFIC RESPONSES
Kidney: Oliguria → ATN (Shock Kidney)
Lung: ARDS
Gut: Mucosal barrier failure → bacterial translocation
Liver: Ischemic hepatitis, ↓ synthetic function
Heart: Myocardial depression
↓
MULTIORGAN DYSFUNCTION SYNDROME (MODS)
↓
DEATH (if untreated)
References: Harrison's Principles of Internal Medicine 22E (2025), Ch. 314, pp. 2353-2360 | Schwartz's Principles of Surgery 11E, Ch. 5, pp. 133-158 | The Essentials of Forensic Medicine & Toxicology 36th Ed. 2026, Ch. 11 (Burns) | Dikshit's Forensic Medicine & Toxicology | Smith & Tanagho's General Urology 19E
add a note on haemorrhagic shock, with forensic aspects
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Loading Image
Loading Image
I now have all the content needed for a comprehensive answer. Here is the detailed note on haemorrhagic shock with forensic aspects:
Haemorrhagic Shock - A Detailed Note with Forensic Aspects
Sources: Harrison's Principles of Internal Medicine 22E (2025); Schwartz's Principles of Surgery 11E; Guyton & Hall Textbook of Medical Physiology 14E; Miller's Anesthesia 10E; Brogdon's Forensic Radiology; DiMaio's Forensic Pathology 3E; The Essentials of Forensic Medicine & Toxicology 36E (2026)
Definition
Haemorrhagic shock is a form of hypovolemic shock caused by acute blood loss resulting in inadequate circulating blood volume, reduced cardiac output, impaired oxygen delivery to tissues, and cellular hypoxia. It is the most common cause of shock in surgical, trauma, and forensic settings.
As Schwartz defines it: "The most common cause of shock in the surgical or trauma patient is loss of circulating volume from haemorrhage."
Normal Blood Volume Reference Points
| Patient | Total Blood Volume |
|---|---|
| Average 70-kg adult male | ~5 litres (70 mL/kg) |
| Adult female | ~65 mL/kg |
| Neonate | ~85 mL/kg |
| Infant | ~80 mL/kg |
- Systolic BP does not fall significantly until >30% (1500 mL) of circulating volume is lost
- A patient may lose up to 750-1000 mL (15%) with minimal clinical signs - "silent" haemorrhage
- Apparent tachycardia and hypotension together represent both significant blood loss AND physiologic decompensation
Pathophysiology of Haemorrhagic Shock
Step 1 - Acute Blood Loss → Initial Haemodynamic Instability
Blood loss → ↓ venous return → ↓ ventricular end-diastolic volume (preload) → ↓ stroke volume → ↓ cardiac output → ↓ arterial pressure
Step 2 - Baroreceptor Activation (Seconds to Minutes)
- Aortic arch and carotid sinus baroreceptors detect ↓ arterial wall stretch → disinhibit vasomotor centres in the brainstem → intense sympathetic outflow
- Atrial low-pressure stretch receptors activated even by mild haemorrhage → earlier alarm signal
- Result: ↑ heart rate, ↑ myocardial contractility, peripheral vasoconstriction (α₁-adrenergic)
Step 3 - Neuroendocrine Cascade (Minutes to Hours)
| Hormone | Source | Action |
|---|---|---|
| Catecholamines (Epi/NE) | Adrenal medulla / sympathetic nerves | ↑ HR, ↑ contractility, vasoconstriction, glycogenolysis |
| Cortisol | Adrenal cortex (via ACTH) | Gluconeogenesis, insulin resistance, Na⁺ retention |
| ADH/Vasopressin | Posterior pituitary | Water retention, vasoconstriction, hepatic gluconeogenesis |
| Renin → Angiotensin II → Aldosterone | Kidney → Liver → Adrenal | Vasoconstriction, Na⁺/H₂O retention |
| Glucagon | Pancreatic α-cells | Glycogenolysis, gluconeogenesis |
| Growth hormone | Anterior pituitary | Protein anabolism, lipolysis |
Net effect: Peripheral vasoconstriction (blood shunted from gut, kidney, skin to heart and brain); salt and water conservation; hyperglycaemia and catabolism.
Step 4 - Cellular Hypoxia and Anaerobic Metabolism
- ↓ O₂ delivery → Anaerobic glycolysis → Lactic acid accumulation
- Only 2 ATP generated per glucose (vs 38 aerobically)
- Lactate rises in serum, muscle, and liver (lactate = marker of oxygen debt severity)
- ATP depletion → failure of Na⁺/K⁺ ATPase → cellular swelling, loss of membrane integrity
- Ca²⁺ influx → phospholipase and protease activation → irreversible cell death
- Metabolic acidosis impairs enzyme activity, coagulation, and myocardial contractility
Step 5 - Microcirculatory Failure
- Sustained vasoconstriction → sludging of RBCs in capillaries, rouleaux formation
- Capillary leak from endothelial swelling → fluid shifts into interstitium → worsens intravascular depletion
- No-reflow phenomenon: Cellular swelling occludes capillaries; perfusion cannot be restored even after macroscopic volume replacement
- DAMPs (HMGB1, mitochondrial DNA, formyl peptides) released from ischemic cells → amplify systemic inflammatory response
Progressive vs Nonprogressive Haemorrhagic Shock (Guyton)
A critical concept from experimental physiology (Guyton, Fig. 24.2):
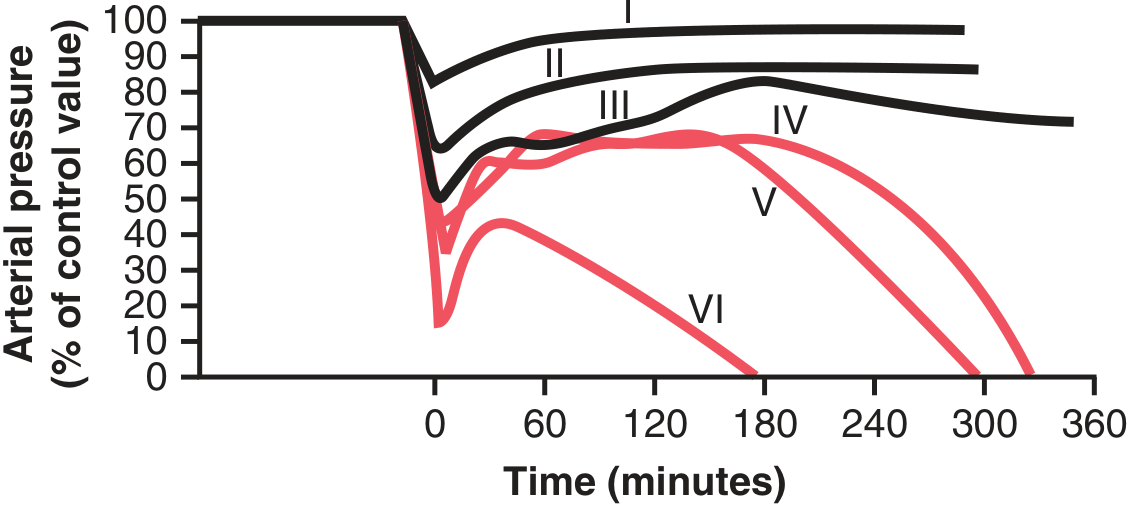
Fig. 24.2 (Guyton & Hall): Time course of arterial pressure after different degrees of acute haemorrhage. Groups I-III recovered; Groups IV-VI entered irreversible shock.
- Nonprogressive (Compensated) shock: Blood loss below a critical threshold; negative feedback mechanisms restore circulation
- Progressive shock: Blood loss exceeds critical threshold (MAP <45 mmHg in experimental models); shock perpetuates itself through positive feedback loops → "vicious cycle of shock"
- Even a few extra mL of blood loss beyond this critical threshold is the difference between survival and death
Positive Feedback Loops Driving Irreversibility (Guyton Fig. 24.3)
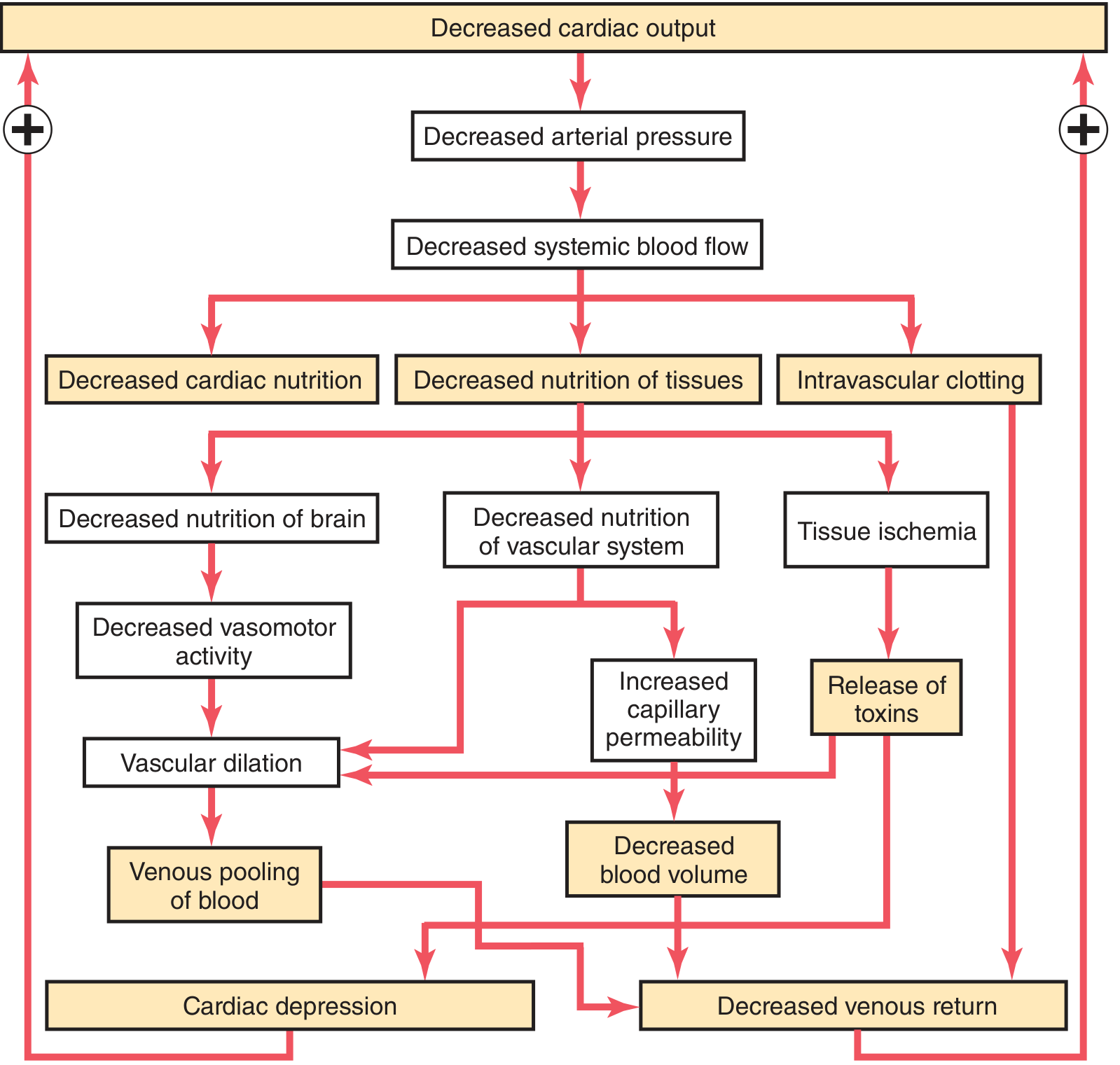
Fig. 24.3 (Guyton & Hall): Positive feedback mechanisms that cause progressive irreversible shock.
The key positive feedback loops are:
- Cardiac depression: ↓ MAP → ↓ coronary perfusion → myocardial ischaemia → ↓ contractility → further ↓ CO. The heart has 300-400% functional reserve, so this manifests only after sustained severe hypotension (usually >2-4 hours)
- Vasomotor failure: Prolonged ischaemia of vasomotor centres in brainstem → sympathetic vasomotor failure → vascular dilation → venous pooling → further ↓ venous return
- Increased capillary permeability: Tissue ischaemia → toxic products → endothelial damage → plasma leak → further ↓ blood volume
- Intravascular clotting (DIC): Stasis + endothelial damage + tissue factor release → microthrombi → block capillaries → further ischaemia; consume clotting factors → bleeding diathesis
- Gut ischaemia → "Toxic factor": Ischaemic bowel → bacterial translocation + endotoxin → systemic inflammation → myocardial depression, further vasodilation
- Cardiac metabolic acidosis: Lactic acid from ischaemic tissues → reduced cardiac contractility (acidosis impairs Ca²⁺ handling in myocardium)
ATLS Classification of Haemorrhagic Shock (Schwartz Table 7-4)
| Class I | Class II | Class III | Class IV | |
|---|---|---|---|---|
| Blood loss (mL) | Up to 750 | 750-1500 | 1500-2000 | >2000 |
| Blood loss (% BV) | Up to 15% | 15-30% | 30-40% | >40% |
| Pulse rate | <100 | >100 | >120 | >140 |
| Blood pressure | Normal | Normal | Decreased | Decreased |
| Pulse pressure | Normal/↑ | Decreased | Decreased | Decreased |
| Respiratory rate | 14-20 | 20-30 | 30-40 | >35 |
| Urine output (mL/h) | >30 | 20-30 | 5-15 | Negligible |
| CNS/Mental status | Slightly anxious | Mildly anxious | Anxious + confused | Confused + lethargic |
Key clinical points:
- BP does not fall until Class III (>30% loss) - tachycardia is the earliest sign
- Pulse pressure narrows in Class II (↑ SVR → ↑ diastolic BP, while systolic is maintained)
- Pregnant patients have expanded blood volume (by ~40%) - must lose more blood before manifesting signs; the fetus may be severely compromised before the mother shows obvious shock
- Elderly on beta-blockers: tachycardia is masked - unreliable sign
Response to Initial Fluid Resuscitation (Schwartz)
Patients categorised as:
- Responders: Haemodynamics normalise and remain stable → likely no significant ongoing haemorrhage
- Transient responders: Initial improvement then re-deterioration → likely ongoing active haemorrhage; require urgent intervention
- Nonresponders: No improvement despite aggressive resuscitation → immediate operative control of haemorrhage required
"Lethal Triad" of Haemorrhagic Shock
In severe haemorrhagic shock, three interacting factors produce a self-perpetuating fatal spiral:
HYPOTHERMIA
/ \
/ \
COAGULOPATHY ←——→ ACIDOSIS
- Hypothermia: Blood/fluid loss + heat redistribution → ↓ core temperature → impairs platelet function, coagulation enzyme activity, myocardial contractility
- Acidosis: Lactic acidosis from anaerobic metabolism → impairs coagulation factor function (especially at pH <7.2), worsens coagulopathy
- Coagulopathy: Trauma-induced coagulopathy (TIC) - dilution from crystalloid resuscitation, factor consumption, fibrinolysis → ongoing uncontrolled haemorrhage
Modern damage control resuscitation targets this triad: permissive hypotension + haemostatic resuscitation (1:1:1 ratio of pRBC:FFP:platelets) + early surgical bleeding control.
Haematological Response to Haemorrhage
| Time Post-Haemorrhage | Change | Mechanism |
|---|---|---|
| 0-30 min | Haematocrit normal | Blood loss is whole blood (no dilution yet) |
| 30 min - 6 hours | Haematocrit begins to fall | Interstitial fluid shifts into intravascular space |
| 6-24 hours | Haematocrit falls further | Continued interstitial-to-intravascular fluid shift |
| 24-72 hours | Reticulocytosis begins | Erythropoietin release stimulates bone marrow |
| 1-2 weeks | Rising RBC count | Bone marrow compensatory erythropoiesis |
Forensic implication: A normal haematocrit in an acute traumatic death does NOT rule out massive haemorrhage - the haematocrit only falls with time, and in immediate death the haematocrit may be completely normal.
FORENSIC ASPECTS OF HAEMORRHAGIC SHOCK
1. Minimum Fatal Blood Loss
| Blood Loss | Consequence |
|---|---|
| Up to 15% (~750 mL) | Compensated; rarely fatal in healthy adult |
| 30-40% (~1500-2000 mL) | Significant shock; may be fatal if untreated |
| >40% (>2000 mL) | Immediately life-threatening; rapid death likely |
| ~50% (~2500 mL) | Rapidly fatal in most cases; death within minutes without intervention |
However, factors modifying fatal blood loss:
- Age (elderly less tolerant)
- Pre-existing cardiac or pulmonary disease
- Speed of blood loss (rapid loss is worse than slow)
- Site of haemorrhage (internal vs external)
- Ambient temperature (cold → vasospasm slows loss)
- Prior anaemia or polycythaemia
2. Autopsy Findings in Death from Haemorrhagic Shock
External Findings
- Pallor of skin, lips, conjunctivae, nail beds
- Pale or absent lividity (livor mortis): Haemorrhagic shock deaths show sparse, faint, or absent livor mortis because blood volume is severely depleted - insufficient blood in skin capillaries to produce the normal hypostatic discolouration. This is a classic forensic marker.
- Wounds, lacerations, stab wounds, or surgical incisions as source of haemorrhage
- Features of internal haemorrhage: abdominal distension, bruising of flanks (Grey-Turner sign in retroperitoneal haemorrhage)
Internal Findings
- Bloodless/pale organs: Heart, lungs, liver, spleen appear pale and anaemic
- Empty or collapsed great vessels (Vanishing Aorta sign - see below)
- Blood pooled externally (in body cavities or at wound site): haemothorax, haemoperitoneum, haemopericardium, retroperitoneal haematoma
- Spleen: Contracted, firm, pale - the spleen contracts to deliver its blood reserve in response to haemorrhage (the normal adult spleen holds ~200-300 mL blood)
- Heart: Pale myocardium; ventricular chambers empty or contain minimal blood; right ventricle may show subendocardial petechiae from air sucked in during agony
- Kidneys: Pale cortex (ischaemia); in survivors who died later - features of shock kidney/ATN (see previous note)
- Brain: Pale, ischaemic changes in prolonged cases; may show watershed infarcts in survivors
The "Vanishing Aorta" Sign (Forensic Radiology)
From Brogdon's Forensic Radiology:
"During fatal haemorrhage, the arteries collapse, which is best seen in big vessels like the aorta, especially the aortic arch and the descending aorta, giving rise to the term 'vanishing aorta'."
- Also seen: collapsed pulmonary arteries and reduced heart chamber sizes on postmortem CT (pmCT)
- Collapsed caval vein is an almost universal postmortem finding → not specific for haemorrhage
- A collapsed IVC with a small heart in the absence of significant dependent decomposition = strongly suggestive of haemorrhagic death
- Postmortem CT is increasingly used in forensic centres: mediastinal haematoma adjacent to aorta = aortic rupture (even without contrast); large haemothorax/haemoperitoneum quantifiable on non-contrast CT
3. Distinguishing Cause of Haemorrhage at Autopsy
| Source | Key Findings |
|---|---|
| Stab wound | Track through organ/vessel; wound dimensions may indicate weapon |
| Gunshot wound | Entry/exit wounds; bullet track; projectile recovery |
| Blunt trauma rupture (liver, spleen, aorta) | Laceration pattern; associated fractures (pelvic fracture → retroperitoneal haematoma) |
| Ruptured aortic aneurysm | Atherosclerotic/dilated aorta; no external trauma |
| Ruptured ectopic pregnancy | Free blood in peritoneum; tubal/ovarian pathology in female |
| Ruptured oesophageal varices | Liver disease; blood in stomach/GI tract |
| Pulmonary haemorrhage | Frothy blood-stained fluid in airways |
4. Timing of Death from Haemorrhage (Forensic Estimation)
| Rate of Haemorrhage | Approximate Time to Death |
|---|---|
| Aortic/carotid transection | Seconds to 2-3 minutes |
| Major vessel laceration (femoral, subclavian) | 3-10 minutes |
| Hepatic/splenic laceration | Minutes to hours |
| Slow internal haemorrhage (e.g., retroperitoneal) | Hours to days |
| Complications (ATN, ARDS, sepsis) | Days to weeks |
Forensic importance of the "lucid interval":
- In slow haemorrhage (e.g., haemopneumothorax, slow retroperitoneal bleed), the individual may remain ambulatory for minutes to hours before collapsing
- This is the "walk-and-die" scenario (analogous to the lucid interval in EDH) - has major medicolegal implications in assault/homicide cases where the victim can describe their assailant before collapse
5. Post-Resuscitation Forensic Findings
Medical intervention before death significantly alters autopsy findings:
- Haemodilution: Resuscitation with crystalloids/colloids dilutes haematocrit → haematocrit at autopsy lower than would be expected from blood loss alone
- Delayed organ failure: Kidneys may show shock nephropathy (ATN); lungs may show ARDS; brain may show watershed infarcts - all date the injury to hours/days before death
- Iatrogenic findings: IV line sites, surgical interventions, drains, intubation - must be documented and distinguished from ante-mortem injury
- Transfusion-related: Massive transfusion → hyperkalaemia, hypocalcaemia, dilutional coagulopathy; may show "transfused" blood of different type in heart chambers
6. Determining Ante-mortem vs Perimortem Haemorrhage
| Feature | Ante-mortem Haemorrhage | Perimortem/Postmortem |
|---|---|---|
| Tissue vital reaction | Present (haemosiderin, macrophages if >24-48h; neutrophil infiltration >6h) | Absent |
| Clot appearance | Organised, firm, adherent; shows layering ("chicken fat" + red cell layer) | Loose, unorganised, non-adherent; may liquify |
| Associated soft tissue | Bruising, swelling, inflammatory infiltrate | No infiltrate |
| Blood volume | Significant pooled blood in cavity = likely ante-mortem | Minimal oozing |
| Biochemistry (vitreous) | Low glucose, elevated lactate = metabolic shock | N/A |
7. Special Forensic Scenarios
Haemorrhage Masquerading as Natural Death
- A person with a previously undiagnosed aortic aneurysm collapses and dies; may be labelled natural death unless autopsy is thorough
- Ruptured splenic artery aneurysm in a young pregnant woman - rare but rapidly fatal; may raise suspicion of foul play without proper investigation
Assault with Delayed Death
- Victim sustains blunt abdominal trauma (kicks, punches) → splenic/hepatic laceration → slow haemorrhage → survives hours before collapsing
- The time between assault and death creates medicolegal questions regarding causation - answered by:
- Wound morphology matching blunt trauma pattern
- Histological dating of haemorrhage (leucocyte infiltration = ante-mortem)
- Documentation of clinical course (hospital records)
Haemorrhage + Other Contributing Factor
- Mild haemorrhage (non-fatal alone) + pre-existing cardiac disease or anticoagulant therapy = death
- The "thin skull" or "eggshell skull" principle in law: an assailant must take their victim as they find them - even if the victim was unusually susceptible, causation is established
DIC in Haemorrhagic Shock
- Massive tissue injury + blood transfusion + prolonged shock → disseminated intravascular coagulation
- Autopsy: microthrombi in glomerular capillaries, adrenal cortex haemorrhage (Waterhouse-Friderichsen pattern), fibrin in pulmonary capillaries
- Amniotic fluid embolism is a special cause of haemorrhagic/obstructive shock + DIC in obstetric fatalities - must be looked for at autopsy (squamous cells and lanugo hairs in pulmonary vasculature)
Management Principles (for exam completeness)
| Priority | Action |
|---|---|
| 1 | Airway + breathing; oxygen |
| 2 | Haemorrhage control - direct pressure, tourniquet, pelvic binder, operative |
| 3 | Two large-bore IV access + send crossmatch, FBC, coagulation, lactate |
| 4 | Permissive hypotension: Target SBP 80-90 mmHg until surgical haemostasis (prevents clot disruption) |
| 5 | Damage control resuscitation: pRBC:FFP:platelets 1:1:1 (haemostatic resuscitation) |
| 6 | Avoid excessive crystalloid (worsens coagulopathy, acidosis, abdominal compartment syndrome) |
| 7 | Tranexamic acid (TXA) within 3 hours of injury (CRASH-2 trial) |
| 8 | Correct lethal triad: warm, correct acidosis, correct coagulopathy |
| 9 | Definitive haemorrhage control: operative or endovascular |
References: Schwartz's Principles of Surgery 11E, Ch. 5 (pp. 133-160) & Ch. 7 (pp. 218-222) | Guyton & Hall Medical Physiology 14E, Ch. 24 (pp. 299-307) | Miller's Anesthesia 10E, Ch. 62 | Harrison's 22E (2025), Ch. 314 | Brogdon's Forensic Radiology, Ch. 26 | The Essentials of Forensic Medicine & Toxicology 36E (2026)
anatomy of heart with blood supply with dissection techniques foramen ovale, CAD and MI for forensic medicne as well Sudden natural death
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Finding Sources
Reading File
Loading Image
Reading File
Loading Image
Reading File
Anatomy of the Heart, Blood Supply, Dissection Techniques, Foramen Ovale, CAD, MI, and Sudden Natural Death
Sources: Gray's Anatomy for Students; Robbins & Kumar Pathologic Basis of Disease; DiMaio's Forensic Pathology 3rd Ed.; The Essentials of Forensic Medicine & Toxicology 36th Ed. (2026); Parikh's Medical Jurisprudence; Fuster & Hurst's The Heart 15th Ed.
PART 1: ANATOMY OF THE HEART
Position and Size
- Located in the middle mediastinum, within the pericardial sac
- Positioned obliquely - two-thirds to the left of the midsternal line
- Approximately 250-350 g in the adult (male heart slightly heavier); heart weighing >350 g in females or >400 g in males is considered pathologically enlarged
- Apex points downward, forward, and to the left - at the 5th left intercostal space, mid-clavicular line
- Base faces posteriorly and superiorly
External Surfaces
- Anterior (sternocostal) surface: Formed mainly by the right ventricle
- Inferior (diaphragmatic) surface: Formed mainly by the left ventricle and small portion of right ventricle
- Posterior (base): Formed mainly by the left atrium
- Left (pulmonary) surface: Formed by the left ventricle
Chambers - Key Anatomical Points
| Chamber | Wall Thickness | Key Internal Features |
|---|---|---|
| Right atrium (RA) | Thin | SVC and IVC openings; coronary sinus opening; fossa ovalis; crista terminalis; pectinate muscles in auricle |
| Right ventricle (RV) | 3-5 mm | Trabeculae carneae; moderator band (septomarginal trabecula); 3-cusp tricuspid valve; infundibulum leading to pulmonary trunk |
| Left atrium (LA) | Thin | 4 pulmonary vein openings; smooth posterior wall; no crista terminalis |
| Left ventricle (LV) | 8-15 mm | Papillary muscles; chordae tendineae; 2-cusp mitral valve; outflow tract → aortic valve |
Cardiac Valves
- Pulmonary valve: 3 semilunar cusps (anterior, left, right) - no papillary muscles/chordae
- Aortic valve: 3 semilunar cusps (left, right, posterior = non-coronary) - the left and right aortic sinuses give off the coronary arteries
- Tricuspid valve: 3 cusps (anterior, posterior, septal)
- Mitral (bicuspid) valve: 2 cusps (anterior/aortic, posterior/mural)
Cardiac Skeleton (Fibrous Skeleton)
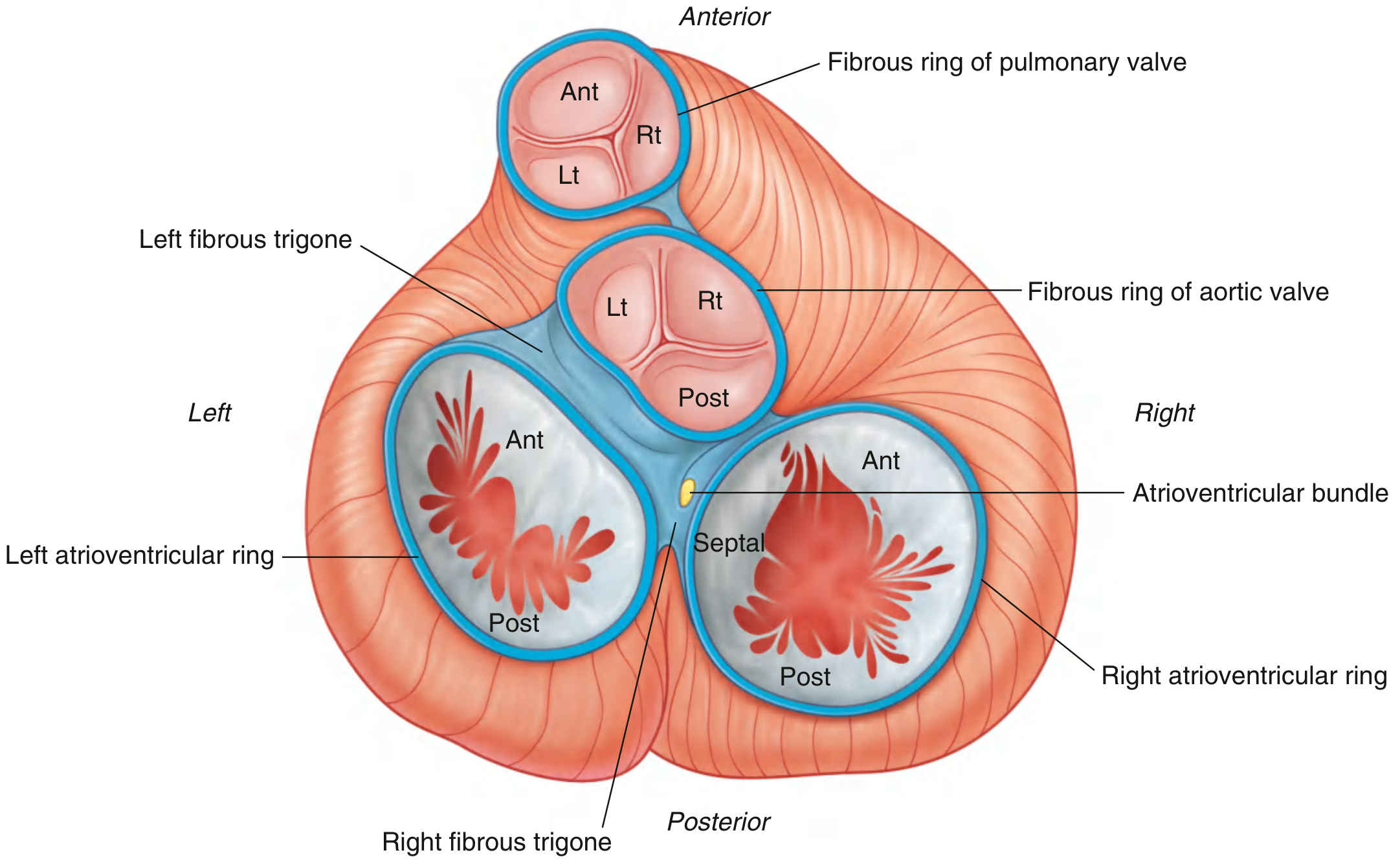
Fig. 3.78 (Gray's Anatomy for Students): Cardiac skeleton - fibrous rings of all four valves, fibrous trigones, and the AV bundle.
The fibrous skeleton consists of:
- Four fibrous valve rings (annuli fibrosi)
- Left and right fibrous trigones connecting the rings
- The right fibrous trigone is the thickest and provides the passage for the Bundle of His (AV bundle)
- Electrically insulates atria from ventricles, forcing conduction through the AV node/Bundle of His
Conduction System
- SA node (sinoatrial node): Situated at junction of SVC and right atrium, supplied by the SA nodal artery (branch of RCA in 60%; LCx in 40%)
- AV node: Situated at the apex of Koch's triangle (triangle of Koch), in the interatrial septum above coronary sinus; supplied by AV nodal artery from RCA (right dominant in 80%)
- Bundle of His: Passes through the right fibrous trigone into the membranous IVS
- Left and Right Bundle Branches
- Purkinje fibres
PART 2: CORONARY ARTERIES (Blood Supply)
(Gray's Anatomy for Students)
Both coronary arteries arise from the aortic sinuses (sinuses of Valsalva) in the initial portion of the ascending aorta, and encircle the heart in the coronary sulcus (atrioventricular groove).
Right Coronary Artery (RCA)
- Origin: Right (anterior) aortic sinus
- Course: Descends in the coronary sulcus between RA and RV → turns posteriorly at the inferior margin → continues on the diaphragmatic surface
Branches:
- SA nodal branch (early atrial branch) - passes posteriorly around the SVC → supplies SA node (in 60% of individuals)
- Right marginal branch - runs along the acute (inferior) margin toward the apex
- AV nodal branch - small branch before the posterior IVS
- Posterior interventricular branch (posterior descending artery - PDA) - runs in the posterior interventricular sulcus toward the apex
Territory supplied by RCA:
- Right atrium and right ventricle
- SA and AV nodes (in right-dominant hearts - 80%)
- Interatrial septum (portion)
- Posterior one-third of interventricular septum
- Inferior/posterior wall of left ventricle (in right-dominant hearts)
Left Coronary Artery (LCA)
- Origin: Left (posterior) aortic sinus
- Course: Passes between pulmonary trunk and left auricle → enters coronary sulcus → immediately divides into two terminal branches
Terminal branches:
1. Left Anterior Descending Artery (LAD) = Anterior interventricular branch
- Passes around left side of pulmonary trunk → descends in the anterior interventricular sulcus toward the apex
- Often continues around the apex onto the posterior surface for a short distance
- Branches: septal perforators (supply anterior 2/3 of interventricular septum), diagonal branches (supply anterior LV surface)
- The "Widow-Maker" - the most critical coronary vessel; proximal LAD occlusion = massive anterior MI
2. Circumflex branch (LCx)
- Courses toward the left in the coronary sulcus onto the diaphragmatic surface
- Gives off left marginal artery across the rounded left border of the heart
- Usually ends before the posterior interventricular sulcus (in right-dominant hearts)
- In left-dominant hearts (~20%): LCx gives off the PDA and supplies the AV node
Territory supplied by LCA:
- Left atrium and most of left ventricle
- Anterior two-thirds of interventricular septum (LAD)
- Anterior wall of LV, apex (LAD)
- Lateral wall of LV (LCx)
Coronary Dominance
| Type | Dominant Artery | PDA Origin | AV Node Supply | Frequency |
|---|---|---|---|---|
| Right dominant | RCA | RCA | RCA | ~80% |
| Left dominant | LCA (LCx) | LCx | LCx | ~15% |
| Co-dominant | Both | Both | - | ~5% |
Venous Drainage
- Coronary sinus (in posterior coronary sulcus) - receives: great cardiac vein, middle cardiac vein, small cardiac vein → empties into RA
- Anterior cardiac veins: Drain directly into RA
- Thebesian veins (venae cordis minimae): Drain directly into cardiac chambers
PART 3: FORAMEN OVALE
Embryological Development
- In fetal life, the foramen ovale (between right and left atria) allows oxygenated blood from the IVC to bypass the non-functioning lungs and pass directly from RA → LA → LV → systemic circulation
- At birth, with the first breath:
- Pulmonary vascular resistance falls → ↑ pulmonary blood flow → ↑ LA pressure
- RA pressure falls (cord clamping → ↑ SVR → ↑ LA return)
- ↑ LA pressure > RA pressure → septum primum is pushed against septum secundum, functionally closing the foramen ovale
- Anatomical fusion occurs gradually over the first year of life
Anatomy of the Fossa Ovalis and Foramen Ovale
- In the adult right atrium, the fossa ovalis is a shallow oval depression on the interatrial septum
- It is the remnant of the foramen ovale
- The limbus fossae ovalis (border of the fossa) is a raised rim - remnant of septum secundum
- The floor of the fossa is formed by the thin septum primum
Patent Foramen Ovale (PFO)
- In approximately 25-30% of adults, the foramen ovale fails to fully fuse anatomically (though functionally closed by LA > RA pressure)
- A probe-patent foramen ovale = the septum is not fused but acts as a one-way valve
- Prevalence: ~27% of adults at autopsy
Forensic Significance of PFO
- Paradoxical embolism: Venous thrombus from deep veins travels to RA → crosses PFO into LA → systemic arterial embolism → stroke in a young person. Forensically: a young person found dead with cerebral infarct and DVT/PE with a PFO demands investigation of paradoxical embolism
- Decompression sickness (divers): Nitrogen bubbles from venous system cross PFO → arterial system → neurological decompression sickness - medicolegal in diving accidents
- Air embolism: In certain trauma scenarios, air entering the venous system can cross a PFO → arterial air embolism → stroke/death
- Emboli in MI: Right-sided emboli (e.g., from right-sided endocarditis or peripheral veins) can cross a PFO and embolise to coronary arteries → MI
- Autopsy documentation: At autopsy, probe patency should always be tested by passing a probe through the fossa ovalis - must be documented in sudden death cases, especially in young individuals with cerebral infarcts or paradoxical emboli
PART 4: CORONARY ARTERY DISEASE (CAD)
Definition and Epidemiology
CAD (Ischemic Heart Disease = IHD) encompasses entities resulting from myocardial ischaemia due to an imbalance between myocardial supply and demand. In >90% of cases, IHD results from obstructive atherosclerotic lesions of epicardial coronary arteries.
- IHD is the single largest cause of mortality worldwide (>15% of global deaths; ~9 million/year in high-resource countries)
- First clinical presentation may be sudden cardiac death (SCD) in ~25% of CAD cases
Pathogenesis of CAD
Atherosclerosis: The primary mechanism.
- Begins in childhood/adolescence as fatty streaks
- Progresses over decades to fibrous plaques → complicated plaques
- Critical stenosis = >70% luminal narrowing → symptomatic on exertion
- 75% narrowing = significant hemodynamic obstruction at rest (DiMaio)
Acute Coronary Syndrome (ACS): Triggered by acute plaque disruption:
- Endothelial erosion or plaque rupture → exposure of subendothelial collagen + necrotic core
- Platelet adhesion → aggregation → release of TXA₂, ADP, serotonin → vasoconstriction
- Coagulation cascade activation (tissue factor) → thrombus formation
- Within minutes: coronary artery may be completely occluded
Forensic Significance of CAD
- 75% of adults dying suddenly and unexpectedly of cardiovascular disease have CAD at autopsy (DiMaio)
- Only 13.4% of sudden CAD deaths show gross acute coronary thrombosis at autopsy - most deaths occur on a background of severe chronic atherosclerosis without acute thrombus
- >75% narrowing of at least one major vessel is present in virtually all sudden CAD deaths
- In most sudden deaths from CAD, at least two vessels are significantly involved
- Single vessel disease: Proximal LAD occlusion ("widow-maker") - most common single vessel cause
Coronary Atherosclerosis Types (Forensic)
| Type | Description |
|---|---|
| Eccentric plaques | Classic asymmetric atherosclerotic narrowing; typical CAD |
| Concentric thickening | Hypertensive cardiovascular disease; uniform wall thickening; lumen appears patent but critically narrowed |
| Calcified rigid tubes | Elderly (>60 yrs); patent but non-compliant; calcified coronary walls |
| Intramural coronary dysplasia | Young patients; severe medial thickening; luminal narrowing of intramural vessels; epicardial arteries appear normal |
Special CAD Entities (Forensic)
- Bridging: A portion of the LAD (most common) courses through the myocardium instead of epicardial fat; systolic compression → reduced diastolic filling at high HR → sudden death with exercise; seen in 30-50% of hypertrophic cardiomyopathy
- Coronary artery spasm: Vasospastic angina; normal coronary arteries at autopsy; death from arrhythmia
- Spontaneous coronary artery dissection (SCAD): 80% in females (especially peripartum); intramural haematoma collapses and occludes lumen; three-quarters involve the LAD; presents as sudden death; changes of cystic medial necrosis may be present
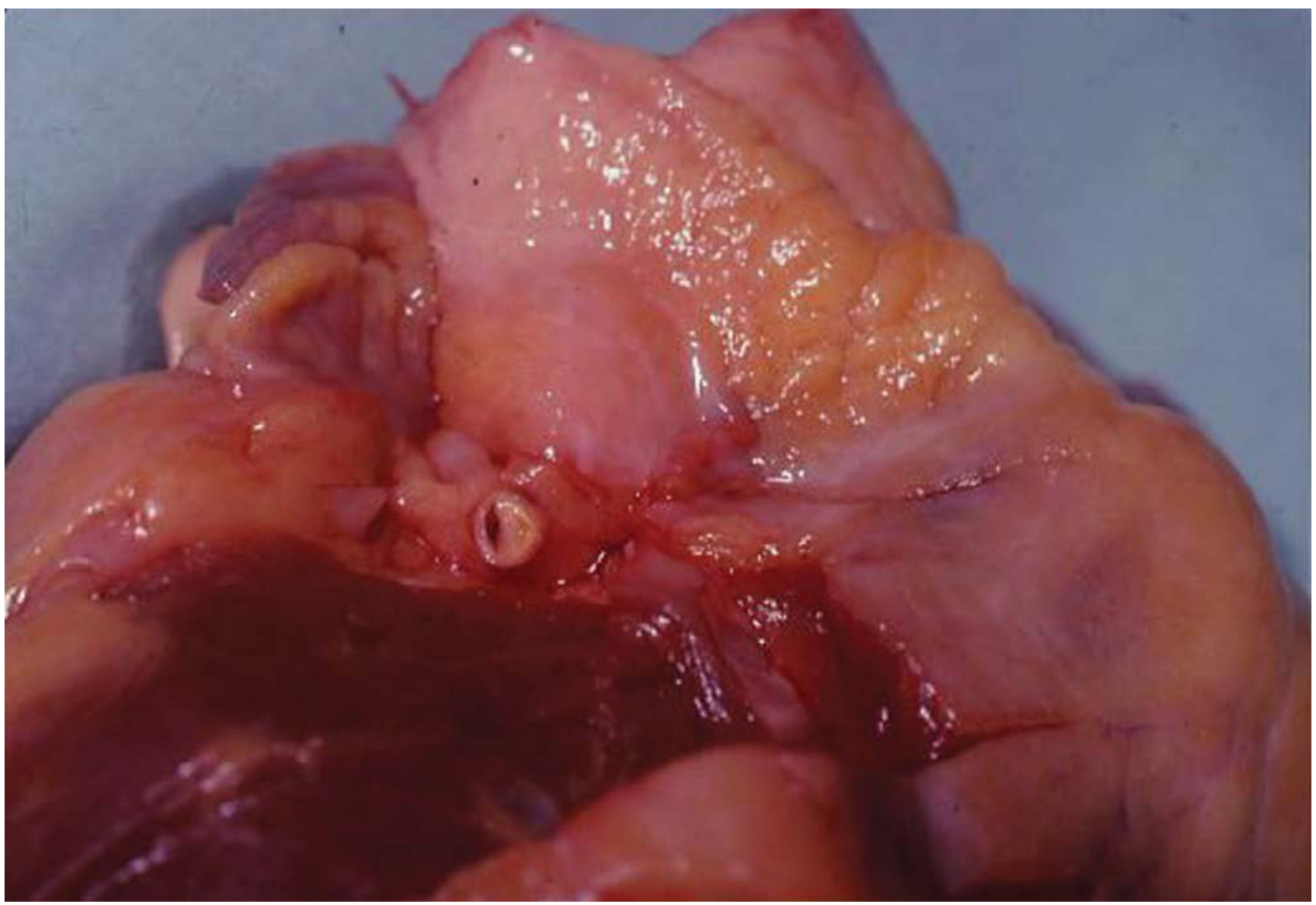
Fig. 3.1 (DiMaio): LAD coronary artery with 75% narrowing of lumen due to atherosclerosis in a 21-year-old male.
PART 5: MYOCARDIAL INFARCTION (MI)
Definition
MI = death of cardiac muscle due to prolonged ischaemia (irreversible necrosis begins after 20-40 minutes of severe ischaemia - blood flow ≤10% of normal).
Critical Timing of Ischaemic Injury (Robbins Table 12.4)
| Event | Time |
|---|---|
| ATP depletion begins | Seconds |
| Loss of contractility | <2 minutes |
| ATP reduced to 50% | 10 minutes |
| ATP reduced to 10% | 40 minutes |
| Irreversible cell injury (necrosis) | 20-40 minutes |
| Microvascular injury | >1 hour |
Distribution of MI by Coronary Artery (Robbins)
| Artery | Frequency | Territory Infarcted |
|---|---|---|
| LAD | 40-50% | Anterior LV wall; apex; anterior 2/3 IVS |
| RCA | 30-40% | Inferior/posterior LV wall; posterior 1/3 IVS; inferior RV free wall |
| LCx | 15-20% | Lateral wall of LV (except apex) |
The LAD, LCx, and RCA collectively cover virtually the entire left ventricle; the right ventricle is uncommonly infarcted in isolation (1-3% of MIs).
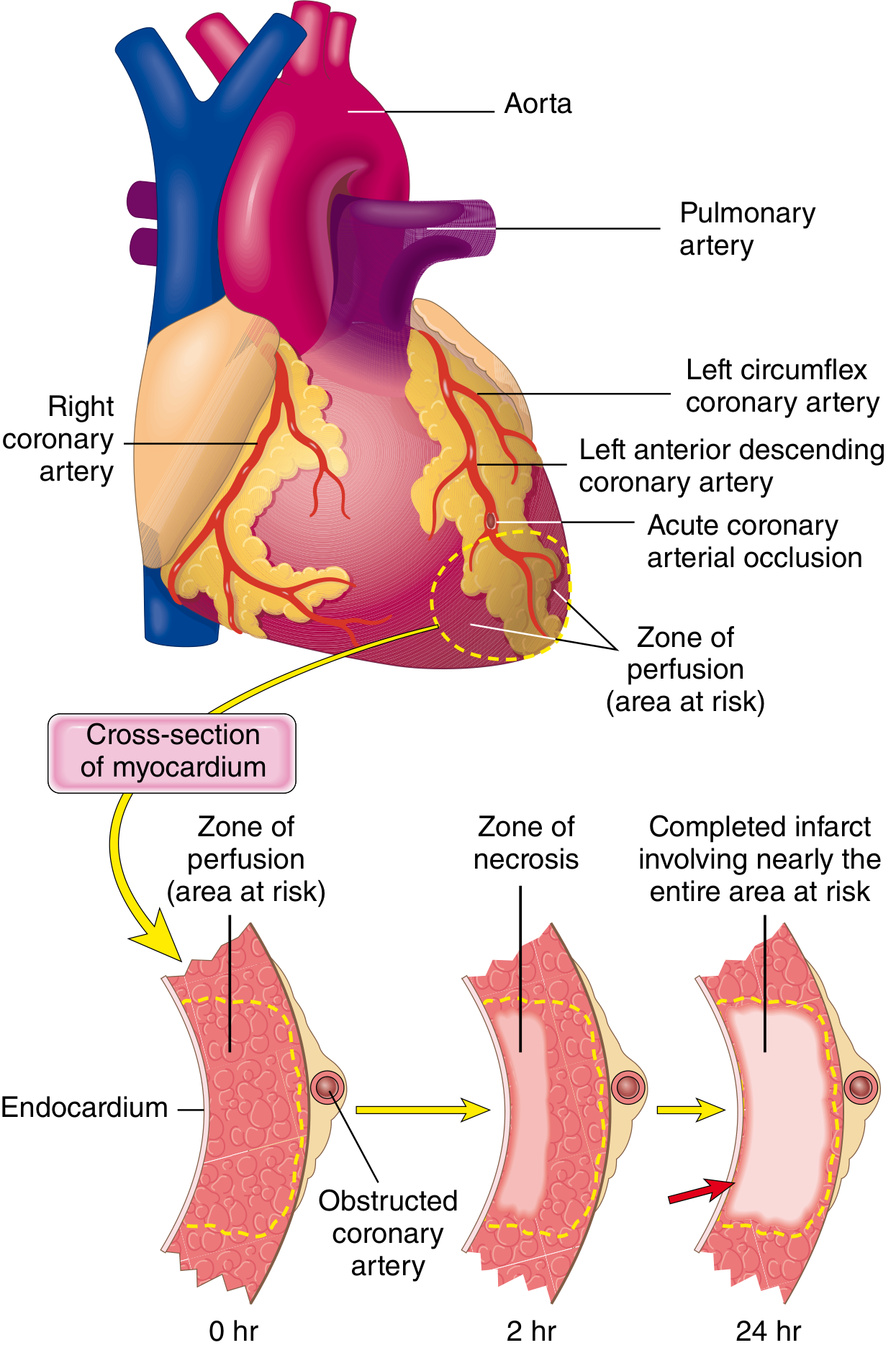
Fig. 12.10 (Robbins): Progression of myocardial necrosis after coronary artery occlusion - the wavefront phenomenon.
Patterns of Infarction
- Transmural MI: Full-thickness necrosis; associated with acute plaque disruption + thrombosis of an epicardial vessel; classic STEMI; a narrow subendocardial rim (~0.1 mm) is spared (diffusion from ventricular cavity)
- Subendocardial (non-transmural) MI: Necrosis confined to inner 1/3-1/2 of wall; due to: thrombus that lyses spontaneously before completing transmural necrosis; or global hypotension/shock → circumferential subendocardial necrosis (watershed ischaemia)
- Multifocal microinfarction: Small intramural vessel pathology (vasculitis, microemboli, cocaine/catecholamine spasm)
Morphological Evolution of MI (Critical Forensic Table)
| Time | Gross Findings | Microscopic Findings |
|---|---|---|
| 0-12 hours | Usually no gross change; TTC staining reveals pale area after 2-3 hours | Early coagulative necrosis; wavy myofibers; "stretched" cells; no inflammatory cells yet |
| 12-24 hours | Pale/mottled area; dark mottling | Neutrophil infiltration begins; coagulative necrosis; loss of nuclei/cross-striations |
| 1-3 days | Hyperaemic border around pale necrotic area | Dense neutrophil infiltration; phagocytosis of dead myocytes beginning |
| 3-7 days | Central yellow-tan softening; hyperaemic border | Macrophage infiltration; granulation tissue at edges; beginning of debris removal |
| 1-3 weeks | Yellow-tan infarct; soft; depressed; fibrotic edges | Granulation tissue replacing necrotic zone; new capillaries; progressive collagen deposition |
| >2 months | White, firm, contracted scar (gelatinous initially, then dense fibrous) | Dense collagenous scar; scanty residual myocytes |
Forensic key: An MI <12 hours old is often invisible at gross autopsy. Always use:
- TTC (Triphenyl Tetrazolium Chloride) staining: Brick-red = viable myocardium (LDH intact); pale/unstained = infarcted myocardium (LDH leaks out). Effective after 2-3 hours post-infarction
- Microscopy is mandatory for MIs <24 hours old
Complications of MI
| Complication | Timing | Notes |
|---|---|---|
| Sudden death/arrhythmia | Immediate to 24h | VF most common; re-entry circuits in ischaemic myocardium |
| Cardiogenic shock | 1-7 days | >40% LV infarction |
| Acute pericarditis | 2-4 days | Fibrinous; pleuritic chest pain |
| Mural thrombus | 1-2 weeks | LA or LV; risk of systemic embolism |
| Myocardial rupture | 3-7 days (peak day 5) | Softening of infarct before adequate collagen formation; haemopericardium + tamponade; forensically: rupture within 1 week of MI = classic finding |
| Papillary muscle rupture | 2-7 days | Acute mitral regurgitation; pulmonary oedema |
| Ventricular aneurysm | Weeks-months | Paradoxical wall motion; mural thrombus |
| Dressler syndrome | 2-10 weeks | Autoimmune pericarditis |
PART 6: DISSECTION TECHNIQUES FOR AUTOPSY EXAMINATION OF THE HEART
Standard Autopsy Techniques (Forensic Medicine)
The heart is examined as part of the medicolegal autopsy using one of four standard dissection techniques:
1. Virchow Technique
- Organs removed one by one and dissected individually
- Most commonly used in medicolegal autopsies
- Each organ is examined, weighed, and sectioned separately
- Advantage: Systematic; thorough examination of each organ
- Disadvantage: Relationships between organs are lost
2. Rokitansky Technique
- In-situ partial dissection: Organs are examined while still in the body; removed in groups
- Thoracic + cervical organs removed as one block; abdominal organs as another; urogenital as a third
- Advantage: Preserves anatomical relationships
- Disadvantage: Less systematic than Virchow
3. Ghon Technique
- Organs removed in groups based on anatomical systems
- Modified variation of Rokitansky
4. Letulle Technique
- En masse removal of all organs (thoracic, cervical, abdominal, pelvic) as one large organ block
- Organ block then dissected on the examination table
- Advantage: Preserves all organ relationships; useful in trauma where injury patterns span multiple systems
- Disadvantage: Time-consuming; less commonly used
Specific Heart Dissection Technique at Autopsy
Step 1 - Removal from chest:
- Heart is removed with a generous portion of great vessels attached (aorta at arch; pulmonary arteries at bifurcation; superior and inferior vena cavae)
Step 2 - External examination:
- Weight (normal: male 250-350 g; female 200-280 g; pathological >400 g male, >350 g female)
- Pericardial sac: fluid amount (normal <50 mL straw-coloured fluid), adhesions, haemopericardium
- Epicardial fat distribution; coronary artery course
- Chamber sizes; external dimensions
Step 3 - Coronary artery examination (CRITICAL):
- The coronary arteries are dissected and serially cross-sectioned at 3-5 mm intervals
- Each major vessel (LAD, LCx, RCA, and proximal branches) is systematically examined
- % luminal narrowing is estimated for each section
- Thrombus, calcification, and haemorrhage within plaques are documented
- Significance of 75% narrowing: Critical threshold for forensic significance (DiMaio)
- In suspected sudden cardiac death: ALL major vessels must be fully serially sectioned - a critical stenosis may be missed with longitudinal cutting
Step 4 - Chamber opening:
- Follow the direction of blood flow ("inflow-outflow" technique):
- Open RA: cut from IVC to auricle → examine tricuspid valve, fossa ovalis, coronary sinus
- Open RV: cut from tricuspid → along diaphragmatic surface → apex → outflow → pulmonary valve
- Open LA: cut between pulmonary veins → examine mitral valve
- Open LV: cut from mitral valve → along left lateral wall → apex
Step 5 - Myocardial sectioning:
- Heart is sliced in short-axis cross-sections (1 cm intervals) from apex to base
- Allows systematic examination of all coronary territories simultaneously
- Look for: areas of pallor (acute ischaemia), yellow/white scarring (old MI), thinning of wall, haemorrhage
Step 6 - Valve examination:
- Circumferences of all four valves measured
- Cusps/leaflets examined for: vegetation, calcification, perforation, prolapse, fusion
Step 7 - Foramen Ovale Examination:
- Probe is passed through the fossa ovalis to test for patent foramen ovale
- If probe passes (probe-patent FO) → document size and note forensic implications
- Must be documented in young sudden death cases
Special Staining for Recent MI at Autopsy
- TTC (Triphenyl Tetrazolium Chloride): Used when MI <24 hours old is suspected; slices immersed in TTC at 37°C → normal myocardium stains brick-red; infarcted area remains pale
- Histology: Essential when MI <12 hours old; wavy fibers and early coagulative necrosis are the earliest findings
PART 7: SUDDEN NATURAL DEATH
Definition
Sudden death is defined as death within 24 hours of onset of symptoms in a person who was apparently in normal health (Parikh). When it is caused entirely by disease, it is sudden natural death. Natural death means that trauma or poison played no part.
- Incidence: approximately 10% of all deaths
System-wise Classification (Essentials of Forensic Medicine & Toxicology 36th Ed., Table 6.1)
| System | % of Sudden Natural Deaths | Major Causes |
|---|---|---|
| Cardiovascular | 45-50% | Coronary atherosclerosis ± thrombosis; hypertensive heart disease; cardiomyopathy; valvular disease; arrhythmias; ruptured aneurysm; pulmonary embolism |
| Respiratory | 15-23% | Lobar pneumonia; bronchopneumonia; TB haemorrhage; pulmonary embolism; asthma; acute glottis oedema; foreign body aspiration |
| CNS | ~15% | Stroke (haemorrhagic/ischaemic); SAH from berry aneurysm; meningitis |
| Gastrointestinal | ~5-8% | Peptic ulcer haemorrhage; ruptured oesophageal varices; acute pancreatitis |
| Genitourinary | ~2-5% | Ruptured ectopic pregnancy; PPH; eclampsia; renal failure |
| Endocrine | ~2% | Diabetic ketoacidosis; Addisonian crisis |
| Infectious | ~2% | Septicaemia; meningococcaemia |
Sudden Cardiac Death (SCD) - Detailed Forensic Analysis
Definition: Unexpected death from cardiac causes within 1 hour of symptom onset (or found dead within 24 hours of last being seen well).
- ~325,000 deaths/year in the USA; 4-5 million worldwide
Mechanism of Death (DiMaio):
- ~80%: Ventricular tachycardia → ventricular fibrillation
- ~20%: Asystole or bradyarrhythmia
- Demonstrated by portable cardiac monitor recordings of individuals who collapsed and died
Timing: Circadian variation - peak incidence 7-9 AM (70% higher than rest of day); attributed to increased sympathetic activity and catecholamines in the morning (Willich et al.). Also associated with strenuous exercise and environmental extremes (heat, cold).
Causes of SCD by Age Group
Age >35 years (adults):
- CAD/Coronary atherosclerosis = 75-80% of all SCD (DiMaio)
- ~50% of individuals with CAD die suddenly
- SCD is the first symptom in ~25% of CAD patients
- Only 13.4% show gross acute coronary thrombosis at autopsy (DiMaio, 500 consecutive cases)
- Most have severe chronic atherosclerosis (at least two vessels, ≥75% narrowing) WITHOUT fresh thrombus
- 80-90% of successfully resuscitated SCD patients show NO enzymatic or ECG evidence of acute MI (Robbins)
- Healed remote MIs are present in ~40% of SCD victims
Age <35 years (young SCD - forensically important):
- Hypertrophic cardiomyopathy (HCM): Most common cause of SCD in young athletes; LV wall >15 mm; asymmetric septal hypertrophy; diastolic dysfunction; associated with bridging
- Anomalous coronary artery origin: ALCAPA (anomalous left coronary artery from pulmonary artery); anomalous origin of LCA from right sinus with inter-arterial course
- Arrhythmogenic right ventricular cardiomyopathy (ARVC): Fibro-fatty replacement of RV myocardium; SCD on exertion
- Long QT syndrome / Brugada syndrome: Channelopathies; structurally normal heart at autopsy - diagnosis requires genetic testing
- Myocarditis: Viral (Coxsackie B most common); inflammatory infiltrate + myocyte necrosis
- Mitral valve prolapse
- Commotio cordis: Sudden chest blow during cardiac vulnerable phase of T-wave → VF; normal heart at autopsy
- Drug-related: Cocaine, methamphetamine - vasospasm, arrhythmia; structurally normal or minimal atherosclerosis
- Wolff-Parkinson-White (WPW): Accessory pathway → rapid conduction → VF
Forensic Autopsy Approach in Suspected SCD
External examination:
- Signs of trauma (rule out homicide/accident)
- Signs of cardiac disease: cyanosis, digital clubbing, xanthelasma
- Signs of struggle or resuscitation (CPR injuries: sternal/rib fractures, lacerations)
Internal examination:
- Pericardium: Effusion (>100 mL = tamponade); haemopericardium (rupture?)
- Heart weight: Record; >400 g (male) or >350 g (female) = cardiomegaly
- Coronary arteries: Serial cross-sections at 3-5 mm intervals; ALL major vessels; document % stenosis
- Myocardium: Serial short-axis cross-sections; TTC staining if <24 hours; histology mandatory
- Valves: Vegetation, calcification, prolapse, dimension
- Foramen ovale: Probe patency test
- Conduction system: Special dissection if no cause found (long strips taken from SA node to AV node to bundle branches)
Ancillary investigations in SCD:
- Toxicology: Blood, urine, vitreous, bile, stomach contents - cocaine, alcohol, drugs
- Histology: From right and left ventricles; septum; papillary muscles; SA and AV nodes; coronary arteries with plaques
- Biochemistry: Troponin from pericardial fluid or vitreous (marker of myocardial injury); post-mortem blood glucose; tryptase (anaphylaxis)
- Genetics: BRUGADA gene panel, LQTS genes, ARVC genes (PKP2, DSG2) for unexplained SCD in young
- Radiology: Whole-body CT (Virtopsy) before autopsy - useful for identifying occult injuries, air embolism, coronary calcification
The "Autopsy-Negative" Sudden Death:
When no structural cause is found:
- Cardiac channelopathy (LQTS, CPVT, Brugada) - molecular autopsy required
- Commotio cordis
- Drug-related arrhythmia (cocaine)
- Hyperkalaemia (vitreous potassium important - K⁺ >14 mmol/L in vitreous = ante-mortem hyperkalaemia)
- Anaphylaxis (blood/vitreous tryptase >10 µg/L)
Summary: CAD, MI, SCD Relationships
ATHEROSCLEROSIS (decades of progression)
↓
Critical Coronary Stenosis (>75%)
↓
┌────────────────────────┐
↓ ↓
STABLE CAD PLAQUE RUPTURE
(Angina pectoris) ↓
Acute Thrombus
↓
┌────────────────────────────┐
↓ ↓
Partial/Transient Complete/Sustained
Occlusion Occlusion
↓ ↓
NSTEMI/UA STEMI
↓ ↓
┌──────────────────────────┐
↓ ↓
Arrhythmia (VF) Myocardial Necrosis
↓ ↓
Sudden Cardiac Death Infarct Complications
References: Gray's Anatomy for Students (pp. 234-245) | Robbins & Kumar Pathologic Basis of Disease (pp. 505-525, Chapter 12) | DiMaio's Forensic Pathology 3rd Ed. (Ch. 3, pp. 55-75) | The Essentials of Forensic Medicine & Toxicology 36th Ed. 2026 (Ch. 6, Table 6.1) | Parikh's Medical Jurisprudence & Toxicology | Fuster & Hurst's The Heart 15th Ed.
brain anatomy blood suppy sudden natural death , berrys aneurism dissection techniques, CVA and intraparenchymal bleed
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Loading Image
Reading File
Reading File
Loading Image
Loading Image
Finding Sources
hanging
Reading File
Hanging forensic medicine aspects, mode manner, autopsy techniques, ipc, bns, cause of death
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Reading File
Loading Image
Your credits are exhausted. You can purchase a top-up or subscribe to a plan to continue using Orris. If you're already subscribed, your daily credits will reset tomorrow.
This is a shared conversation. Sign in to Orris to start your own chat.