explain the routes of metastasis and Describe the role of carcinogenic agents and their cellular interactions.
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
brf
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
explain the routes of metastasis and Describe the role of carcinogenic agents and their cellular interactions.
Reading File
Finding Sources
Finding Sources
Searching the Web
routes of metastasis cancer spread mechanisms carcinogenic agents cellular interactions
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Loading Image
Here is a comprehensive, textbook-grounded explanation of both topics.
Routes of Metastasis
Metastasis is defined as the spread of a tumor to sites physically discontinuous with the primary tumor — the defining feature of malignancy. Approximately 30% of newly diagnosed solid tumor patients present with clinically evident metastases, and a further 20% have occult metastases at diagnosis. About 90% of cancer deaths result from metastatic disease, not the primary tumor.
Malignant neoplasms disseminate by three principal pathways:
1. Seeding Within Body Cavities
Occurs when a tumor invades and penetrates the wall of a body cavity, after which cells implant and grow on peritoneal, pleural, pericardial, or subarachnoid surfaces.
- Ovarian carcinoma is the classic example: tumor cells shed into the peritoneal cavity and cover peritoneal surfaces widely, yet may not deeply invade underlying tissues. Here, the ability to implant is separable from invasive capacity.
- CNS tumors (e.g., medulloblastoma, ependymoma) penetrate the cerebral ventricles and are carried by cerebrospinal fluid to implant on meningeal surfaces around the brain or spinal cord.
2. Lymphatic Spread
Lymphatic spread is the most typical route for carcinomas (epithelial tumors).
- The pattern of lymph node involvement follows local lymphatic drainage from the primary site:
- Lung carcinomas → bronchial → tracheobronchial → hilar nodes
- Breast carcinoma (upper outer quadrant) → axillary nodes; medial lesions drain to internal mammary nodes → then supraclavicular/infraclavicular nodes
- "Skip metastases": Cancer cells may bypass the immediately proximal nodes and be trapped in more distal nodes.
- Cells can traverse all nodes and reach the vascular compartment via the thoracic duct.
- Sentinel lymph node: The first node to receive lymph drainage from the primary tumor. Identified using dye or radiolabeled tracers, its biopsy determines extent of spread and guides therapy.
- Enlarged regional nodes do not always mean metastatic involvement — reactive hyperplasia from tumor antigens and necrotic cells can cause lymphadenitis. Biopsy is required for confirmation.
3. Hematogenous Spread
This is the favored route for sarcomas (mesenchymal tumors), though carcinomas use it too.
- Cancer cells penetrate thin-walled veins preferentially over arteries.
- Bloodborne cells arrest in the first capillary bed encountered:
- Portal drainage → liver (most common site)
- Caval drainage → lungs (second most common)
- Tumors near the vertebral column (thyroid, prostate) embolize via the paravertebral venous plexus → preferential spread to the spine.
- Renal cell carcinoma and hepatocellular carcinoma characteristically grow within veins in a snakelike pattern, sometimes extending up the inferior vena cava to the right heart — yet this may not produce widespread dissemination.
- Organ-specific homing patterns:
- Prostatic carcinoma → bone
- Bronchogenic carcinoma → adrenal glands and brain
- Neuroblastoma → liver and bones
- Uveal melanoma → liver
- Skeletal muscle, despite being rich in capillaries, is rarely a metastatic site

Molecular Mechanisms Enabling Metastasis
Epithelial-to-Mesenchymal Transition (EMT)
A key cellular event permitting invasion and metastasis. Carcinoma cells:
- Downregulate epithelial markers (e.g., E-cadherin)
- Upregulate mesenchymal markers (e.g., vimentin, smooth muscle actin)
- Shift morphologically from polygonal epithelioid to spindly mesenchymal shape
- Increase production of proteolytic enzymes to degrade extracellular matrix
The master transcription factors SNAIL and TWIST are key drivers — they repress E-cadherin, breaking cell-cell adhesion. Once cells arrive at distant sites, the reverse process (mesenchymal-to-epithelial transition, MET) allows re-establishment.
Deterministic vs. Probabilistic Models
There is ongoing debate:
- Probabilistic model: As tumor grows, cells randomly accumulate all mutations needed for metastasis — a function of cell number and time
- Deterministic model: Some tumors acquire metastatic mutations early in carcinogenesis, making metastasis an intrinsic tumor property
Carcinogenic Agents and Their Cellular Interactions
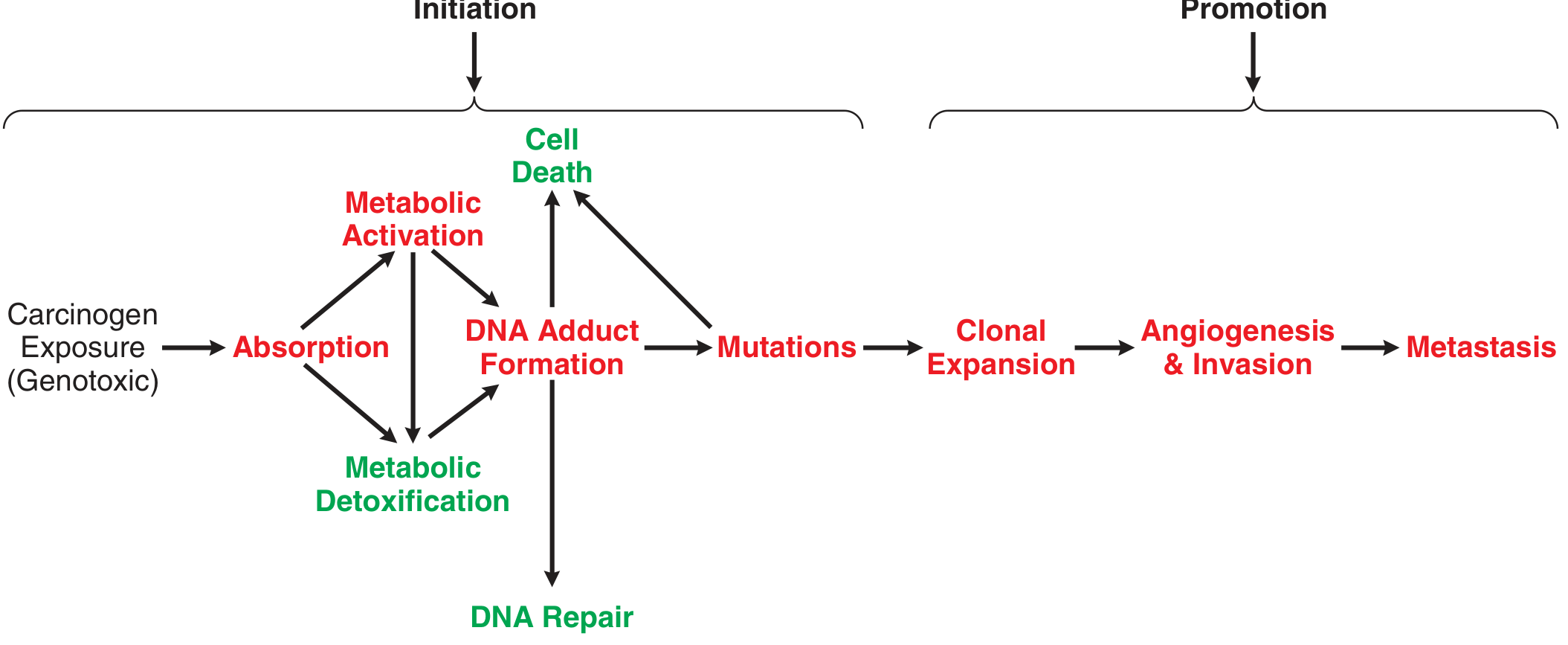
The Multistage Model of Chemical Carcinogenesis
Transformation of a normal cell into malignancy is a multistage process. The classic model involves:
- Initiation — an initiator causes gene mutations that increase proliferative potential and reduce apoptosis (permanent, irreversible)
- Promotion — a promoter does not directly mutate genes but alters signaling pathways or the extracellular environment to increase survival, proliferation, or invasiveness of pre-cancerous cells (reversible if exposure stops)
- Progression — chromosomal instability and accumulating mutations lead to full invasiveness and metastasis
IARC Classification of Carcinogens
The International Agency for Research on Cancer (IARC) classifies compounds into:
| Group | Meaning |
|---|---|
| 1 | Known human carcinogens |
| 2A | Probably carcinogenic |
| 2B | Possibly carcinogenic |
| 3 | Insufficient data |
| 4 | Unlikely to be carcinogenic |
Genotoxic Carcinogens — Direct DNA Damage
These agents initiate tumor formation through direct DNA damage:
- Metabolic activation: Most genotoxic carcinogens are procarcinogens — they undergo metabolism (usually by cytochrome P450 enzymes) in target tissues to form a reactive intermediate (the proximate carcinogen)
- DNA adduct formation: The reactive intermediate covalently bonds to DNA
- Alternatively: Reactive oxygen species (ROS) generated can oxidize DNA or form lipid peroxidation products that react with DNA
- If unrepaired before DNA replication → mutation in a proto-oncogene, tumor suppressor gene, or DNA repair gene → selective growth advantage → cancer
Example: Benzo[a]pyrene (tobacco smoke)
- Oxidized by CYPs → 7,8-dihydrodiol (proximate carcinogen)
- Further oxidized → diol epoxide → reacts directly with DNA
- Or oxidized by aldo-keto reductases → catechol → redox cycles → generates ROS
| Carcinogen Class | Example | Source | Mechanism |
|---|---|---|---|
| Polycyclic aromatic hydrocarbons | Benzo[a]pyrene | Tobacco smoke, charbroiled food | DNA adducts + ROS |
| Nitrosamines | NNK | Tobacco products | Metabolic activation → DNA adducts |
| Aromatic amines | 2-naphthylamine | Dyes, rubber | CYP activation → DNA adducts |
Nongenotoxic Carcinogens — Tumor Promotion Without DNA Damage
These agents increase cancer incidence without directly damaging DNA, mostly acting as tumor promoters:
- Phorbol esters: Mimic diacylglycerol, activate protein kinase C (PKC) isoforms → stimulate MAPK pathways → proliferation, invasiveness, angiogenesis. Normal cells would undergo apoptosis with prolonged activation, but cells with prior mutations resist cell death.
- Estrogenic carcinogens: Activate estrogen receptor alpha (ERα) → stimulate proliferation and invasiveness of estrogen-responsive cells
- Chronic inflammation (e.g., asbestos, hepatotoxic chemicals): Inflammatory cytokines stimulate PKC signaling → proliferation and invasion; compensatory cell cycling during tissue repair increases mutation probability
Epigenetic Mechanisms
Some carcinogens cause heritable changes without DNA sequence alteration:
- Hypermethylation of CpG islands in tumor suppressor gene promoters → silencing
- Demethylation of proto-oncogene promoters → overexpression
- Heavy metals (e.g., arsenic, cadmium): Do not directly damage DNA but interfere with proteins involved in DNA synthesis and repair → increased replication errors
Complete Carcinogens and Cocarcinogens
- Complete carcinogens (e.g., benzo[a]pyrene, UV radiation): Can cause both initiation and promotion with repeated exposure
- Cocarcinogens: Enhance the carcinogenicity of other agents by:
- Increasing absorption (e.g., ethanol acts as a solvent, increasing absorption of tobacco carcinogens in head/neck)
- Depleting protective molecules (ethanol depletes glutathione, impairing detoxification of reactive metabolites)
- Interfering with DNA repair, increasing mutation rates from a second genotoxic agent
Summary Table
| Feature | Genotoxic Carcinogens | Nongenotoxic Carcinogens |
|---|---|---|
| DNA damage | Direct (adducts, strand breaks) | Indirect/none |
| Role | Initiators | Mostly promoters |
| Reversibility | Irreversible mutations | Often reversible if exposure stops |
| Examples | PAHs, nitrosamines, UV, aflatoxin | Phorbol esters, estrogens, asbestos |
| Cellular interaction | Covalent DNA binding, ROS generation | Receptor activation, PKC, cytokines |
Sources:
- Robbins & Kumar Basic Pathology, Chapter 6 — Neoplasia
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, Chapter 76 — Carcinogenesis
Describe the role of carcinogenic agents and their cellular interactions. and Describe the types of hereditary disorders and briefly explain common Mandelian disorders.
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a comprehensive, textbook-based answer on both topics.
PART 1: Role of Carcinogenic Agents and Their Cellular Interactions
The Multistage Model of Chemical Carcinogenesis
Transformation of a normal cell into malignancy is a multistage process. Exogenous chemicals can act at one or more stages. The classic framework consists of three sequential stages:
| Stage | Agent | Cellular Effect |
|---|---|---|
| Initiation | Initiator | Causes permanent gene mutations → increased proliferation, reduced apoptosis |
| Promotion | Promoter | Alters signaling pathways or extracellular environment → increased survival, proliferation, invasiveness of pre-cancerous cells (no direct DNA damage; reversible if exposure stops) |
| Progression | Continued insults | Chromosomal instability + accumulating mutations → full invasiveness and metastasis |
IARC Classification of Carcinogens
| Group | Classification |
|---|---|
| 1 | Known human carcinogens |
| 2A | Probably carcinogenic in humans |
| 2B | Possibly carcinogenic in humans |
| 3 | Inadequate data |
| 4 | Unlikely to be carcinogenic |
Category 1: Genotoxic Carcinogens — Direct DNA Damage
These agents initiate tumor formation through DNA damage. Most are procarcinogens that require metabolic activation before becoming reactive.
Cellular Interaction Sequence:
- Absorption → carcinogen enters target tissue
- Metabolic activation by cytochrome P450 (CYP) enzymes → proximate carcinogen (reactive intermediate)
- DNA adduct formation — the reactive intermediate covalently bonds to DNA bases
- Alternatively → reactive oxygen species (ROS) generated → oxidize DNA or form lipid peroxidation products that react with DNA
- If unrepaired before DNA replication → permanent mutation in:
- Proto-oncogene → gain of function, uncontrolled proliferation
- Tumor suppressor gene → loss of brake on growth
- DNA repair gene → increased future mutation rate
- Mutated cell with selective growth advantage → clonal expansion → cancer
Prototype Example: Benzo[a]pyrene (tobacco smoke)
- Oxidized by CYPs → 7,8-dihydrodiol (proximate carcinogen)
- Second oxidation by CYP → diol epoxide → covalently reacts with DNA
- Alternatively, oxidized by aldo-keto reductases → catechol → redox cycles → generates ROS
Key Genotoxic Carcinogens Table
| Class | Example | Source | Mechanism |
|---|---|---|---|
| Polycyclic aromatic hydrocarbons (PAHs) | Benzo[a]pyrene | Tobacco smoke, charbroiled food, fossil fuel combustion | Metabolic activation → DNA adducts + ROS |
| Nitrosamines | NNK (nicotine-derived nitrosaminoketone) | Tobacco products | Metabolic activation → DNA adducts |
| Aromatic amines | 2-naphthylamine | Dyes, rubber manufacturing | CYP activation → DNA adducts |
| Aflatoxin B1 | Aflatoxin | Mold on peanuts/grains | Epoxide formation → DNA adducts, p53 mutation |
| Alkylating agents | Cyclophosphamide | Chemotherapy | Direct alkylation of DNA bases |
Category 2: Nongenotoxic Carcinogens — Tumor Promotion Without DNA Damage
These agents increase cancer incidence without directly damaging DNA. They act primarily as tumor promoters by modifying cell signaling.
Mechanisms of Cellular Interaction:
A. PKC/MAPK Pathway Activation (Phorbol esters)
- Phorbol esters mimic diacylglycerol (DAG), activating protein kinase C (PKC) isoforms
- PKC activates mitogen-activated protein kinase (MAPK) pathways → stimulates proliferation, invasiveness, and angiogenesis
- Normal cells would undergo apoptosis with prolonged MAPK activation, but cells with prior apoptosis-suppressing mutations are resistant
B. Estrogen Receptor Activation
- Estrogenic carcinogens bind estrogen receptor alpha (ERα) → stimulate proliferation and invasiveness of estrogen-responsive cells (e.g., breast, endometrium)
C. Chronic Inflammation
- Inflammatory cytokines → stimulate PKC signaling → proliferation, invasion, angiogenesis
- Examples: asbestos (causes mesothelioma via persistent pleural inflammation), chronic hepatotoxic chemicals (stimulate compensatory liver cell proliferation → increases mutation probability)
D. Epigenetic Mechanisms
- Heavy metals (arsenic, cadmium, nickel): do not directly damage DNA but interfere with DNA synthesis and repair proteins → increased replication errors
- Hypermethylation of CpG island promoters → silencing of tumor suppressor genes
- Demethylation of proto-oncogene promoters → overexpression
- These epigenetic changes are heritable through cell division
Category 3: Complete Carcinogens and Cocarcinogens
- Complete carcinogens (e.g., benzo[a]pyrene, UV radiation): can drive both initiation and promotion with repeated exposure
- Cocarcinogens enhance carcinogenicity of other agents by:
- Increasing absorption (ethanol acts as a solvent, increasing tobacco carcinogen absorption in head/neck mucosa)
- Depleting protective molecules (ethanol depletes glutathione/GSH, impairing detoxification of reactive metabolites and ROS)
- Interfering with DNA repair, amplifying mutations from a second genotoxic agent
Summary: Genotoxic vs. Nongenotoxic
| Feature | Genotoxic | Nongenotoxic |
|---|---|---|
| DNA damage | Direct (adducts, strand breaks, oxidation) | Indirect or none |
| Role in carcinogenesis | Initiators | Mostly promoters |
| Reversibility | Irreversible mutations | Often reversible |
| Examples | PAHs, nitrosamines, UV, aflatoxin | Phorbol esters, estrogens, asbestos |
| Key cellular target | DNA bases, tumor suppressors, oncogenes | Receptors (PKC, ERα), cytokine pathways |
PART 2: Types of Hereditary Disorders and Common Mendelian Disorders
Hereditary disorders are classified into four broad categories:
Types of Hereditary Disorders
| Type | Mechanism | Examples |
|---|---|---|
| 1. Mendelian (Single-Gene) Disorders | Mutation in a single gene; follows Mendelian inheritance | Cystic fibrosis, Marfan syndrome, PKU |
| 2. Chromosomal Disorders | Numeric or structural abnormalities of chromosomes | Down syndrome (Trisomy 21), Turner syndrome, Klinefelter syndrome |
| 3. Complex Multigenic Disorders | Polygenic + environmental interactions | Diabetes mellitus, hypertension, coronary artery disease |
| 4. Single-Gene Disorders with Atypical Inheritance | Triplet repeats, mitochondrial inheritance, genomic imprinting | Fragile X syndrome, Huntington disease, Prader-Willi syndrome |
Mendelian (Single-Gene) Disorders — Inheritance Patterns
Single-gene mutations follow three inheritance patterns:
A. Autosomal Dominant (AD)
- Manifested in the heterozygous state
- One affected parent → 50% chance of transmission to each child
- Both sexes affected and transmit equally
- New mutations can occur (no affected parent)
- Features: reduced penetrance (carrier is phenotypically normal), variable expressivity (same mutation, different severity)
- Involved genes usually encode regulatory proteins, receptors, structural proteins — not enzymes (since 50% enzyme activity is often sufficient)
- Age of onset often delayed (e.g., Huntington disease)
B. Autosomal Recessive (AR)
- Manifested only in the homozygous state
- Both parents are typically unaffected carriers
- Each child of two carriers has a 25% chance of being affected
- More often involve enzyme deficiencies — 50% enzyme activity from one normal allele is usually sufficient
- Often more severe and uniform in presentation than AD disorders
- Consanguinity increases risk
C. X-Linked Disorders
- Gene is on the X chromosome
- Males (hemizygous XY) are almost always affected
- Females (XX) are usually carriers with mild or no symptoms (one normal X provides protection)
- Father-to-son transmission does not occur
- Examples: Duchenne muscular dystrophy, Hemophilia A and B, Glucose-6-phosphate dehydrogenase deficiency
Important caveats:
- A single gene mutation may have many phenotypic effects (pleiotropy) — e.g., Marfan syndrome affects skeleton, eyes, and cardiovascular system from one fibrillin gene mutation
- Different mutations can cause the same phenotype (genetic heterogeneity) — e.g., retinitis pigmentosa
- Modifier genes at other loci can alter severity (as seen in cystic fibrosis)
Estimated Prevalence of Common Mendelian Disorders
| Disorder | Inheritance | Prevalence |
|---|---|---|
| Familial hypercholesterolemia | AD | 1 in 500 |
| Polycystic kidney disease | AD | 1 in 1,000 |
| Marfan syndrome | AD | 1 in 5,000 |
| Huntington disease | AD | 1 in 10,000 |
| Sickle cell anemia | AR | 1 in 500 (U.S. African descent) |
| Cystic fibrosis | AR | 1 in 3,200 (North European descent) |
| Tay-Sachs disease | AR | 1 in 3,500 (Ashkenazi Jewish) |
| Phenylketonuria (PKU) | AR | 1 in 10,000 |
| Duchenne muscular dystrophy | X-linked | 1 in 3,500 (males) |
| Hemophilia | X-linked | 1 in 5,000 (males) |
Common Mendelian Disorders — Brief Descriptions
1. Marfan Syndrome (AD)
- Gene/Protein: Mutation in FBN1 gene → defective fibrillin-1 (extracellular matrix glycoprotein)
- Fibrillin forms microfibrils that provide structural support in connective tissue and regulate TGF-β signaling
- Features (pleiotropy from one gene):
- Skeletal: Tall stature, long extremities (arachnodactyly), pectus deformities, scoliosis, joint laxity
- Ocular: Ectopia lentis (displaced lens), myopia
- Cardiovascular: Aortic root dilation → aortic regurgitation, aortic dissection/rupture; mitral valve prolapse
2. Ehlers-Danlos Syndromes (EDS) (AD or AR depending on subtype)
- Gene/Protein: Mutations affecting collagen synthesis or processing
- Subtypes:
- Vascular EDS: COL3A1 mutation → deficient type III collagen → arterial and bowel rupture (AD)
- Kyphoscoliotic EDS: Lysyl hydroxylase deficiency → defective collagen crosslinks → congenital scoliosis + ocular fragility (AR)
- Classical EDS: COL5A1/COL5A2 mutations → deficient type V collagen (AD)
- Features: Hyperflexible joints (predisposition to dislocation), hyperextensible and fragile skin, internal organ rupture
3. Familial Hypercholesterolemia (AD)
- Gene/Protein: Loss-of-function mutations in LDL receptor gene (80–85% of cases)
- Normal function: LDL receptors in hepatocytes bind and internalize LDL, providing feedback to suppress intracellular cholesterol synthesis (via HMG-CoA reductase suppression)
- Defect: Fewer/absent functional LDL receptors → LDL not cleared from plasma → elevated circulating LDL → loss of feedback control → continued cholesterol synthesis
- Features:
- Premature atherosclerosis and myocardial infarction (MI may occur before age 20 in homozygotes)
- Xanthomas (cholesterol deposits in tendons, skin)
- Xanthelasmas (periorbital deposits)
- Corneal arcus
- Heterozygotes: LDL ~2× normal; Homozygotes: LDL ~5× normal (severe, often fatal in childhood)
4. Cystic Fibrosis (AR)
- Gene/Protein: Mutations in CFTR gene → defective/absent cystic fibrosis transmembrane conductance regulator (a chloride ion channel)
- Most common: ΔF508 deletion (deletion of phenylalanine at position 508)
- Defect: CFTR cannot be properly folded → retained in ER → degraded; absent Cl⁻ secretion → thick, viscous secretions in exocrine glands
- Features:
- Lungs: Chronic pulmonary infections (Pseudomonas aeruginosa, Staphylococcus aureus), bronchiectasis, progressive respiratory failure
- Pancreas: Exocrine pancreatic insufficiency → malabsorption, steatorrhea
- Reproductive: Male infertility (absent vas deferens); reduced female fertility
- Sweat glands: Elevated sweat Cl⁻ (diagnostic: sweat chloride > 60 mEq/L)
- Modifier genes at other loci affect severity
5. Phenylketonuria — PKU (AR)
- Gene/Protein: Deficiency of phenylalanine hydroxylase (PAH) → inability to convert phenylalanine to tyrosine
- Defect: Phenylalanine accumulates → toxic to developing brain neurons
- Features:
- Progressive intellectual disability (if untreated)
- Seizures, behavioral issues
- Fair skin and hair (tyrosine needed for melanin synthesis)
- Musty odor (phenylketones in urine/sweat)
- Management: Phenylalanine-restricted diet from birth (newborn screening); detected by Guthrie test
6. Sickle Cell Anemia (AR)
- Gene/Protein: Point mutation in HBB gene → glutamic acid → valine substitution at position 6 of β-globin → hemoglobin S (HbS)
- Under low O₂, HbS polymerizes → sickle-shaped rigid red cells
- Features: Hemolytic anemia, vaso-occlusive crises (bone pain, acute chest syndrome), splenic sequestration, stroke, organ damage
- Carriers (sickle cell trait) are protected against severe malaria — explains high allele frequency in malaria-endemic regions
7. Familial Hypercholesterolemia vs. Related Enzyme Disorders (AR examples)
Gaucher Disease (most common lysosomal storage disease):
- Glucocerebrosidase deficiency → glucocerebroside accumulates in macrophages (liver, spleen, bone marrow)
Tay-Sachs Disease:
- Hexosaminidase A deficiency → GM2 ganglioside accumulates in neurons → progressive neurodegeneration, death by age 2–5 (classic infantile form)
8. Huntington Disease (AD) — Atypical: Triplet Repeat
- Gene: CAG trinucleotide repeat expansion in HTT gene → toxic gain-of-function of huntingtin protein
- Normal: < 36 repeats; Disease: > 40 repeats
- Anticipation: Repeat expands in successive generations → earlier onset and greater severity in offspring
- Features: Progressive chorea, personality changes, dementia; onset typically 30–50 years; uniformly fatal
- Mechanism: Mutant huntingtin forms nuclear inclusions → disrupts transcription, causes neuronal apoptosis (particularly striatum and cortex)
Key Distinctions Between Types
| Feature | AD | AR | X-Linked |
|---|---|---|---|
| Copies needed for disease | 1 (heterozygous) | 2 (homozygous) | 1 (males) |
| Unaffected carriers | Rare (low penetrance) | Both parents typically | Females (carriers) |
| New mutations | Common | Rare | Common |
| Protein type affected | Regulatory, structural, receptors | Enzymes, channels | Variable |
| Father-to-son transmission | Yes | Yes | No |
Sources:
- Robbins & Kumar Basic Pathology, Chapter 4 — Genetic and Pediatric Diseases
- Goodman & Gilman's The Pharmacological Basis of Therapeutics, Chapter 76 — Carcinogenesis
explain with detail in easy language
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.