Nephrotic syndrom as a md pathology resident
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Searching PubMed
"nephrotic syndrome" AND "glomerular disease" AND pathology
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
Loading Image
Finding Sources
Finding Sources
Reading File
Loading Image
I now have all the content needed. Let me compile the comprehensive pathology-resident-level answer.
Nephrotic Syndrome - Pathology Resident Review
Sources: Robbins & Kumar Basic Pathology; NKF Primer on Kidney Diseases, 8e; Brenner & Rector's The Kidney; Frameworks for Internal Medicine
Definition & Diagnostic Criteria
Nephrotic syndrome is a clinical syndrome defined by:
- Proteinuria >3.5 g/24 hours (or spot protein:creatinine ratio >3.5)
- Serum albumin <2.5-3.5 g/dL (hypoalbuminemia)
- Hyperlipidemia (total cholesterol usually >180 mg/dL; elevated LDL, VLDL; reduced HDL)
- Lipiduria (oval fat bodies, fatty casts - "Maltese cross" pattern under polarized light)
- Edema (anasarca, periorbital, dependent)
The unifying pathological mechanism is loss of glomerular charge and size selectivity, maintained normally by the GBM, fenestrated endothelium, and especially podocytes (visceral epithelial cells). The common final morphologic pathway is podocyte foot process effacement on electron microscopy.
Pathophysiology of Complications
| Complication | Mechanism |
|---|---|
| Edema | Hypoalbuminemia → ↓ oncotic pressure → fluid extravasation; + Na retention via RAAS activation |
| Hyperlipidemia | ↓ oncotic pressure stimulates hepatic lipoprotein synthesis; ↓ lipoprotein lipase |
| Lipiduria | Lipoprotein leakage across damaged GBM |
| Hypercoagulability | Urinary loss of antithrombin III, proteins C and S, plasminogen; hepatic overproduction of fibrinogen → renal vein thrombosis (especially in membranous nephropathy and amyloidosis); risk highest when proteinuria >10 g/day + albumin <2 g/dL |
| Infections | Urinary loss of IgG, reduced complement activity, impaired T-cell function → cellulitis, peritonitis (Streptococcus pneumoniae in children) |
| Iron-deficiency anemia | Loss of transferrin |
| Vitamin D deficiency | Loss of vitamin D-binding protein |
| AKI | Low effective circulating volume due to ↓ oncotic pressure |
Primary (Idiopathic) Causes - The "Big 3" for Adults
1. Minimal Change Disease (MCD)
Epidemiology: Most common cause in children (70-90%); ~15% of adult nephrotic syndrome. Peak age 1-7 years.
Associations: Usually idiopathic. Secondary associations include NSAIDs, Hodgkin lymphoma (paraneoplastic - suggestive of T-cell mediated mechanism), thymoma.
Pathogenesis: A circulating permeability factor (likely T-cell derived) injures podocytes causing selective loss of slit diaphragm function. Exact molecule remains uncharacterized. The response to steroids strongly implicates immune dysregulation.
Morphology:
| Modality | Finding |
|---|---|
| Light microscopy | Normal glomeruli (hence "minimal change"). Proximal tubular cells show protein droplets and lipid vacuoles ("lipoid nephrosis") |
| Immunofluorescence | Negative - no immune deposits |
| Electron microscopy | Diffuse effacement of podocyte foot processes (the only abnormality) + podocyte vacuolization, microvillus formation, occasional focal detachments |
Fig. 12.4A - MCD: Normal glomerulus on PAS stain
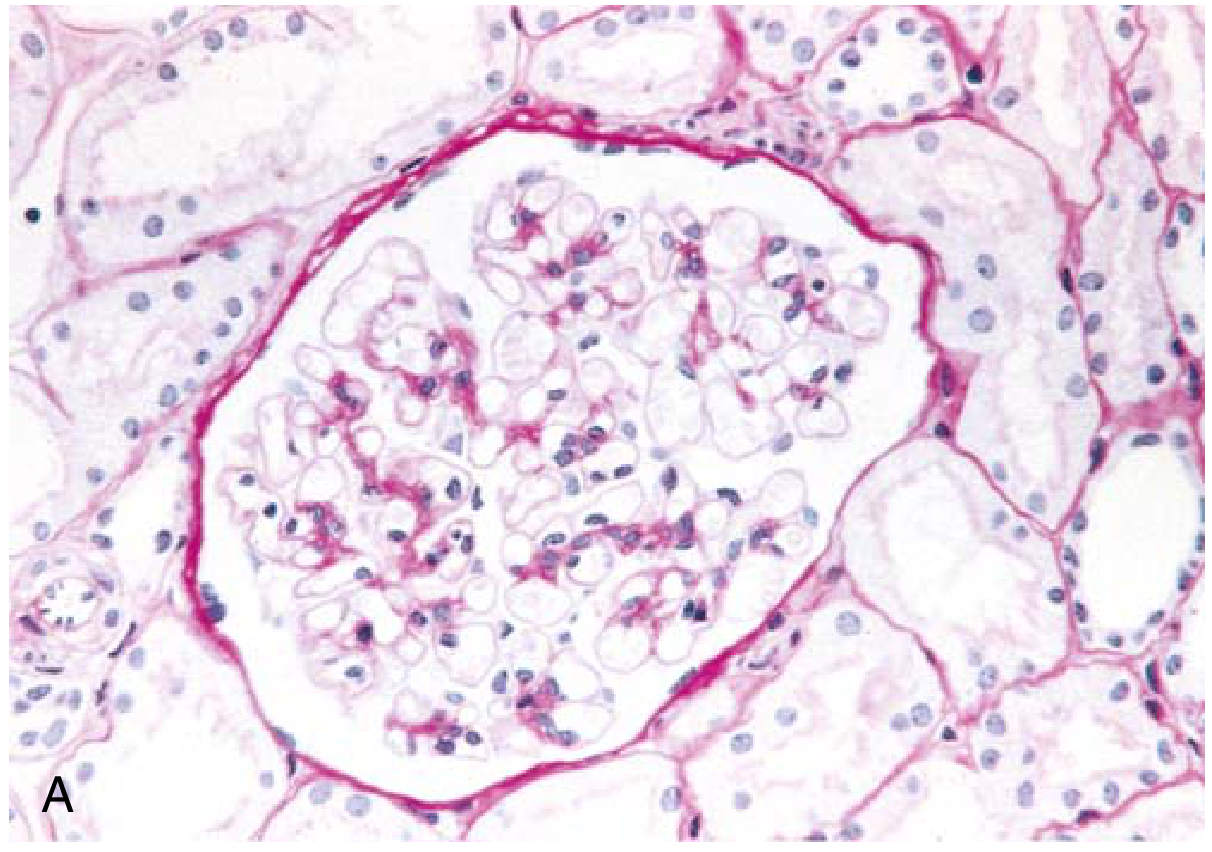
Fig. 12.4B - MCD: EM showing foot process effacement (arrows), CL = capillary lumen, E = epithelial cell, M = mesangium
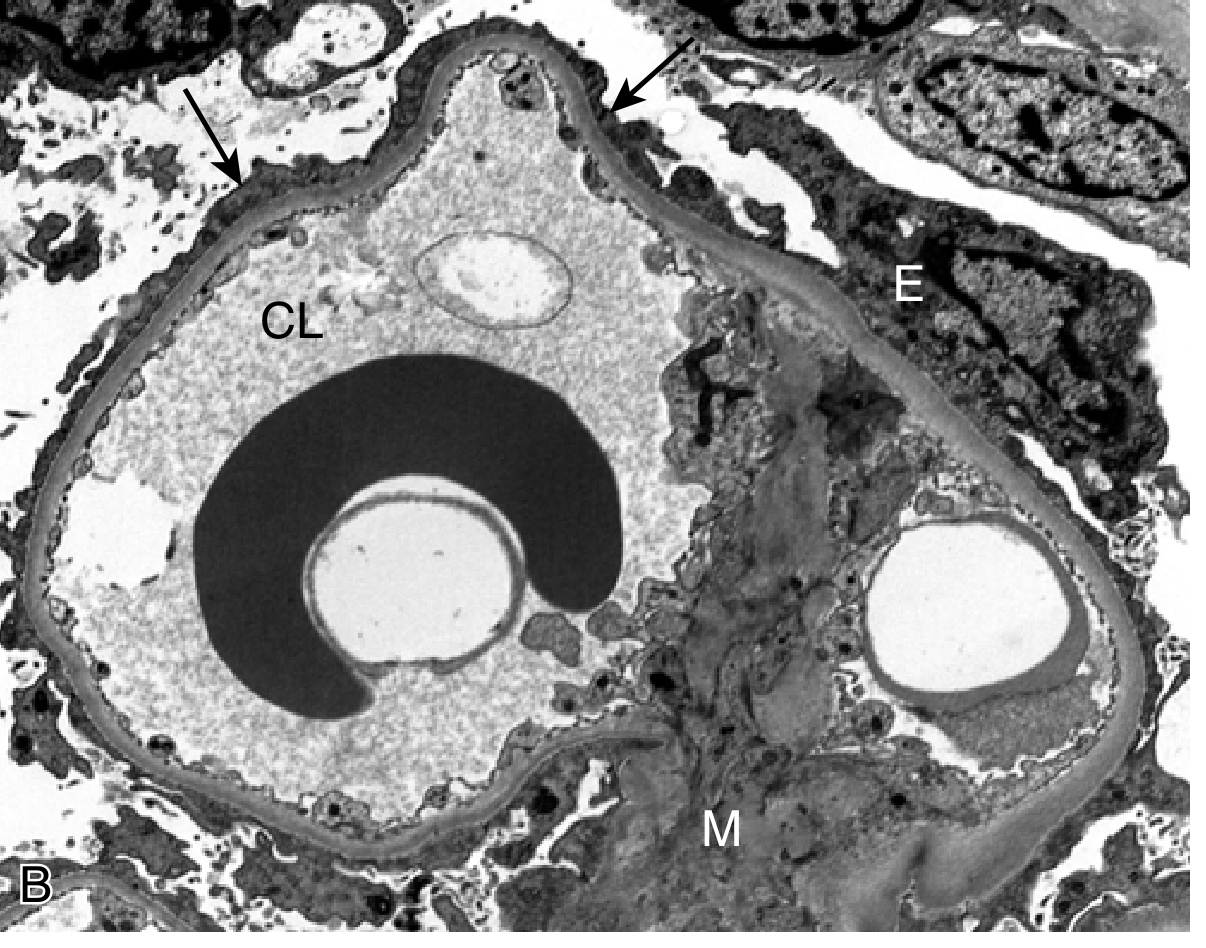
Clinical features: Abrupt-onset nephrotic syndrome; selective proteinuria (mainly albumin - selectivity index <0.1); preserved renal function. >90% of children respond to corticosteroids. Relapses occur in >2/3 of initial responders; some become steroid-dependent. <5% progress to CKD; most often evolve to FSGS. In adults, biopsy is mandatory.
2. Focal Segmental Glomerulosclerosis (FSGS)
Definition: A pattern of injury (not a single disease) - sclerosis of some (focal, <50%) glomeruli involving only a segment of each affected glomerulus.
Epidemiology: ~30% of adult nephrotic syndrome in the US; the most common cause in African Americans. Most common glomerular disorder progressing to ESRD. Less common in children (<15%).
Etiologic Classification:
| Type | Mechanism/Notes |
|---|---|
| Primary (idiopathic) | Circulating permeability factor (lymphocyte-derived); recurs after transplant within hours-days |
| Secondary - maladaptive | Nephron loss from any cause → hyperfiltration in residual glomeruli (obesity, solitary kidney, reflux nephropathy, surgical reduction) |
| Secondary - infections | HIV (HIV-associated nephropathy - HIVAN; collapsing variant) - incidence declining with ART; heroin use |
| Secondary - genetic | Autosomal recessive; >60 genes implicated - nephrin (NPHS1), podocin (NPHS2), alpha-actinin-4, TRPC6; steroid-resistant forms in children |
Pathogenesis: Podocyte injury is the initiating event. Podocyte foot process fusion → ↑ glomerular permeability → podocyte loss → loss of slit diaphragms → ECM deposition → obliteration of capillaries → glomerulosclerosis. Residual podocytes undergo hypertrophy but cannot adequately cover the GBM.
Morphology:
| Modality | Finding |
|---|---|
| Light microscopy | Focal (initially juxtamedullary) and segmental glomerular involvement; obliterated capillary lumina, increased mesangial matrix, hyaline deposits (plasma protein insudation), foamy macrophages; tubular atrophy and interstitial fibrosis in advanced disease |
| Immunofluorescence | Nonspecific IgM and C3 trapping in areas of sclerosis |
| Electron microscopy | Diffuse foot process effacement (may be focal in secondary forms) |
Fig. 12.5 - FSGS: A) Focal sclerosis in one of three glomeruli; B) Segmental sclerosis with hyaline deposit (arrow)
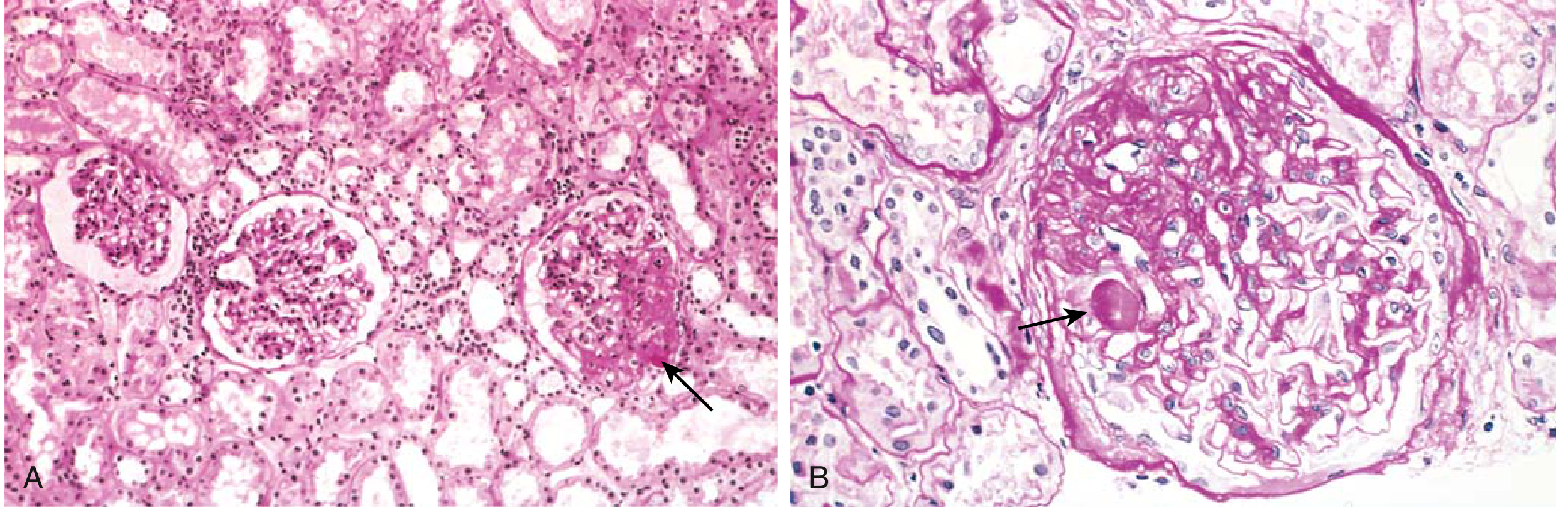
FSGS Morphologic Variants (Columbia Classification):
| Variant | Key Feature | Clinical Significance |
|---|---|---|
| NOS (not otherwise specified) | Most common; classic segmental sclerosis | Variable prognosis |
| Perihilar | Sclerosis at vascular pole | Common in maladaptive/secondary; better prognosis |
| Cellular | Endocapillary hypercellularity, foam cells, epithelial hyperplasia | May represent early tip or collapsing |
| Tip lesion | Sclerosis at tubular pole (opposite vascular pole) | Better prognosis; often steroid-responsive |
| Collapsing | Global/segmental tuft collapse + overlying podocyte hypertrophy and hyperplasia | WORST prognosis; strongly associated with HIV (HIVAN), COVID-19 (COVAN), APOL1 risk variants in Black patients |
Clinical: Nephrotic proteinuria often non-selective. More hematuria and hypertension than MCD. Steroid response poor compared to MCD. Majority progress to ESRD. Recurrence in transplant in primary FSGS (50% within 1-2 years) suggests circulating mediator.
3. Membranous Nephropathy (MN)
Epidemiology: Most common cause of nephrotic syndrome in white adults (30-60 years). Approximately 80% primary, 20% secondary.
Pathogenesis (Primary): Autoimmune - autoantibodies form in situ immune complexes with podocyte surface antigens:
- Anti-PLA2R (anti-phospholipase A2 receptor): present in 70-80% of primary MN; IgG4 subclass; serologic titer useful for diagnosis AND monitoring treatment response
- Anti-THSD7A (thrombospondin type 1 domain-containing 7A): ~3-5%, associated with cancer
- Other antigens: NELL-1, Sema3B, EXT1/EXT2 (lupus-associated)
Secondary causes:
- Infections: hepatitis B (common in Asia), syphilis, malaria, schistosomiasis
- Malignancy: carcinomas (lung, colon, breast), CLL, lymphoma
- Autoimmune: SLE (Class V lupus nephritis), rheumatoid arthritis
- Drugs: penicillamine, gold, NSAIDs, captopril
Complement activation: Despite IgG4 being a poor classical pathway activator, complement proteins are detected (possibly via lectin pathway or IgG1/IgG3 minor components).
Morphology:
| Modality | Finding |
|---|---|
| Light microscopy | Diffuse thickening of GBM without proliferation or inflammation (silver stain shows "spikes" projecting from GBM into subepithelial space between deposits) |
| Immunofluorescence | Granular deposits of IgG (IgG4) and C3 along GBM in a peripheral capillary loop pattern |
| Electron microscopy | Subepithelial electron-dense deposits + GBM "spike and dome" pattern + diffuse foot process effacement. Progressive disease: deposits incorporated into GBM → GBM thickening |
Fig. 12.6 - Membranous Nephropathy: A) GBM thickening (silver stain); B) Granular IgG IF along GBM; C) Subepithelial deposits (arrow), foot process effacement
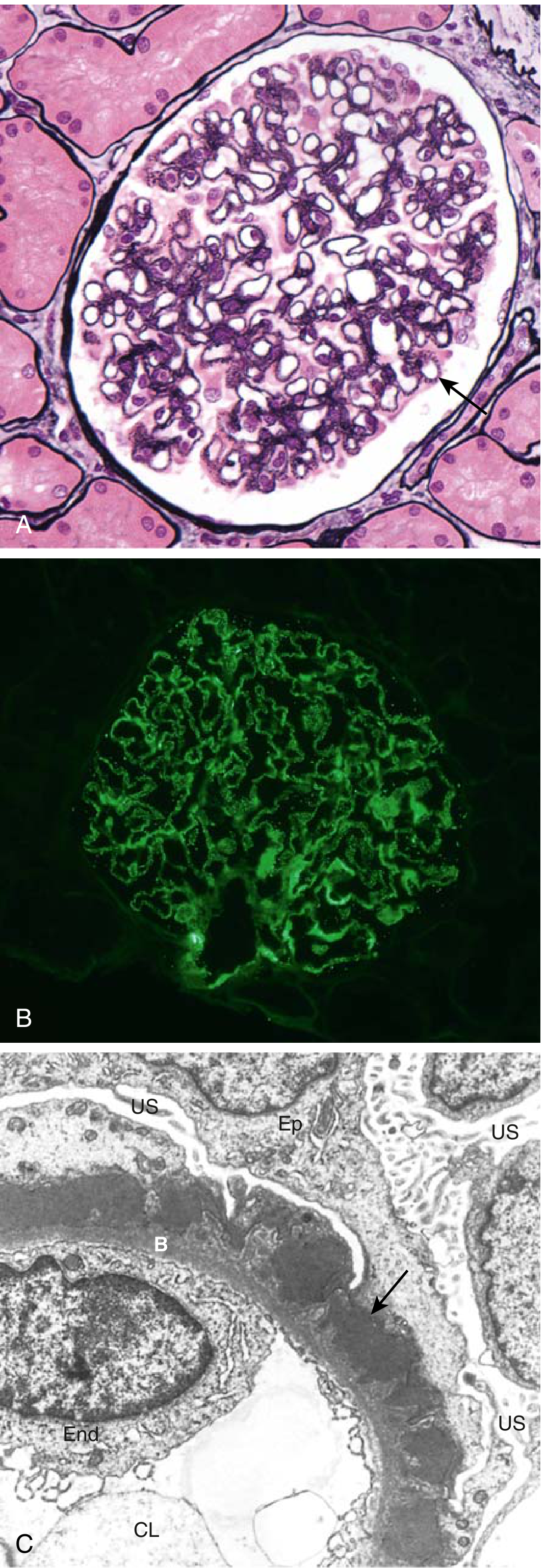
Staging (Ehrenreich-Churg): Stage I (small subepithelial deposits) → Stage II (spikes) → Stage III (deposits incorporated into GBM) → Stage IV (GBM irregular thickening, obsolescent glomeruli)
Clinical: Proteinuria is non-selective (large proteins lost). Does NOT respond to corticosteroids (unlike MCD). Highly variable course - "rule of thirds": 1/3 spontaneous remission, 1/3 persistent proteinuria without progression, 1/3 progressive renal failure. Highest risk of renal vein thrombosis among all nephrotic causes.
4. Membranoproliferative Glomerulonephritis (MPGN)
Definition: Pattern of glomerular injury with GBM thickening + mesangial hypercellularity. Accounts for 5-10% of idiopathic nephrotic syndrome in children and adults. May present with mixed nephrotic-nephritic picture.
Classification (current):
| Type | Mechanism | IF Pattern | EM |
|---|---|---|---|
| Immune complex-mediated MPGN (old "Type I") | Circulating IC or in situ complex formation; activate classical and alternative complement | Granular IgG + C3 in mesangium and capillary walls | Subendothelial + mesangial deposits |
| Complement-mediated (C3 glomerulopathy) | Dysregulation of alternative complement pathway (C3 nephritic factor, CFH mutation); no IC | C3 only (minimal Ig) | Dense intramembranous deposits (dense deposit disease / old "Type II") or subendothelial deposits (C3 GN) |
Light microscopy (MPGN):
- Lobular accentuation of glomerular architecture
- Mesangial and endocapillary proliferation + infiltrating leukocytes
- GBM thickening with "tram-track" / double-contour appearance (due to mesangial cell interposition and new GBM layer)
Fig. 12.7 - MPGN: A) Mesangial proliferation, GBM splitting (silver stain); B) Granular IgG + C3 by IF; C) Subendothelial deposits (arrow) and mesangial deposits
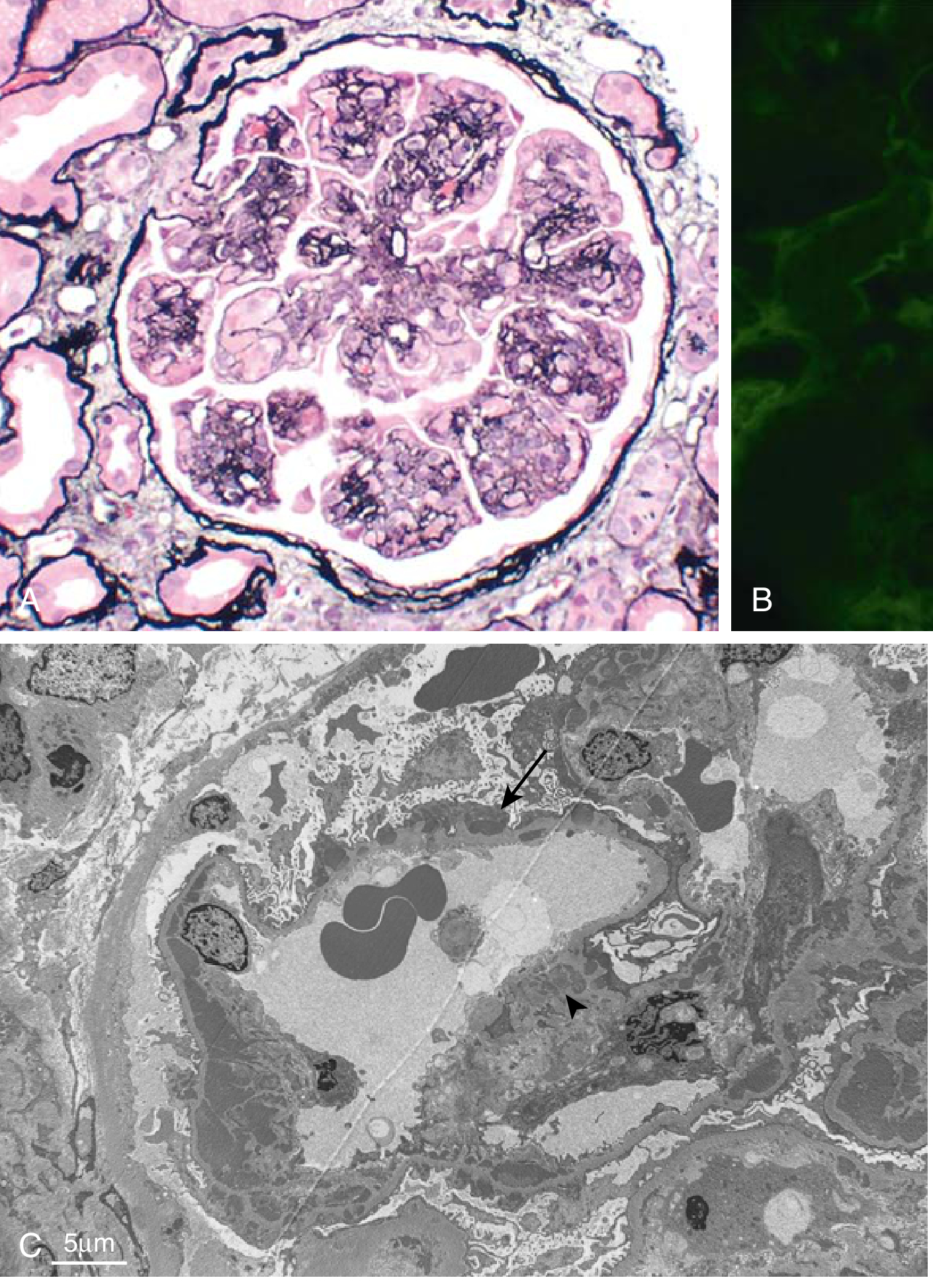
Clinical: Slowly progressive. Presents in adolescents/young adults. Associated with hepatitis C (cryoglobulinemia) in adults - test all adults with MPGN for HCV. Treatment of underlying cause may resolve secondary MPGN.
Secondary / Systemic Causes
Diabetic Nephropathy (Diabetic Kidney Disease)
Pathogenesis: Hyperglycemia → non-enzymatic glycosylation + advanced glycation end-products (AGEs) → GBM thickening, mesangial expansion. Hemodynamic changes (hyperfiltration, ↑ glomerular pressure) contribute via RAAS and TGF-β activation.
Morphology:
- GBM thickening (earliest finding on EM)
- Diffuse mesangial expansion (most common LM finding)
- Nodular glomerulosclerosis = Kimmelstiel-Wilson nodules - PAS+ ovoid nodules in periphery of glomerulus (pathognomonic when present; ~30% of diabetic nephropathy cases)
- Hyalinosis of afferent AND efferent arterioles (efferent arteriolar hyalinosis = highly specific for DM)
- "Capsular drop" lesion (hyaline material between parietal epithelium and Bowman's capsule)
- "Fibrin cap" lesion (homogeneous eosinophilic material overlying capillary loops)
- IF: IgG, albumin nonspecific linear GBM staining ("insudation")
- EM: GBM thickening, diffuse foot process effacement
Amyloidosis
AL amyloidosis: Monoclonal light chain deposition; kidney involvement most common (proteinuria, nephrotic syndrome); EM shows randomly oriented non-branching fibrils 8-10 nm diameter; Congo red stain positive with apple-green birefringence under polarized light.
AA amyloidosis: Secondary to chronic inflammatory conditions; similar renal morphology. Nephrotic syndrome due to amyloidosis carries highest risk of renal vein thrombosis. Median survival ~16 months when NS is present due to AL amyloid.
Comparison Table: The "Big 4" Primary Causes
| Feature | MCD | FSGS | Membranous | MPGN |
|---|---|---|---|---|
| Peak age | Children (1-7 yr) | Adults (Black > White) | Adults (30-60 yr) | Adolescents/Young adults |
| LM | Normal | Focal-segmental sclerosis | GBM thickening (no proliferation) | Lobular, mesangial proliferation, tram-track |
| IF | Negative | IgM, C3 (nonspecific) | Granular IgG4 + C3 (subepithelial) | Granular IgG + C3 (mesangial + subendothelial) |
| EM | Diffuse FPE only | Diffuse FPE + segmental sclerosis | Subepithelial deposits + spikes + FPE | Subendothelial deposits + GBM duplication |
| Deposit location | None | None (nonspecific) | Subepithelial | Subendothelial (± mesangial) |
| Key antibody | None characterized | None (circulating factor) | Anti-PLA2R (70-80%) | None (or IC related to HBV/HCV) |
| Proteinuria selectivity | Selective (albumin) | Non-selective | Non-selective | Non-selective (mixed nephrotic-nephritic) |
| Steroid response | Excellent (>90% children) | Poor | No response | No benefit |
| Progression to ESRD | <5% | Frequent (most common GN→ESRD) | ~40% over 2-20 yr | Slowly progressive |
| Special feature | Lipoid nephrosis; tubular lipid droplets | Collapsing variant = worst Px; APOL1 risk in Black patients | "Spike and dome"; anti-PLA2R monitoring | "Tram-track" GBM; C3 GN variant |
Renal Biopsy Interpretation Framework
For a pathology resident approaching a nephrotic biopsy, evaluate systematically:
-
Light microscopy (H&E, PAS, Silver, Trichrome):
- Cellularity? (proliferative → think MPGN, lupus)
- GBM changes? (thickening → membranous or MPGN; spikes on silver → membranous)
- Sclerosis pattern? (focal-segmental → FSGS; global → end-stage)
- Mesangial expansion? (nodular with PAS+ → diabetic; amyloid → Congo red)
- Tubular changes? (lipid/protein droplets → MCD; atrophy → chronicity)
-
Immunofluorescence:
- IF negative + EM foot process effacement only → MCD
- IF positive → determine pattern and location:
- Granular subepithelial → membranous
- Granular mesangial + subendothelial → MPGN/lupus
- IgA dominant mesangial → IgA nephropathy (nephritic > nephrotic)
- C3 only → C3 glomerulopathy
- Nonspecific IgM/C3 in sclerotic areas → FSGS
-
Electron microscopy:
- Location of deposits (subepithelial / intramembranous / subendothelial / mesangial)
- Degree and distribution of foot process effacement
- Fibril diameter (amyloid ~8-10 nm; fibrillary GN ~12-24 nm; immunotactoid >30 nm)
- GBM thickness (normal: 250-400 nm in women, 300-450 nm in men)
Key Serologic Workup
| Test | Interpretation |
|---|---|
| Anti-PLA2R antibodies | Positive in 70-80% primary MN; titer correlates with activity |
| ANA, anti-dsDNA, C3/C4 | Lupus nephritis (Class V = membranous pattern) |
| SPEP/UPEP + serum free light chains | Amyloidosis, monoclonal gammopathy |
| HBsAg, anti-HCV | Membranous (HBV), MPGN (HCV) |
| HIV | FSGS (collapsing) |
| Blood glucose, HbA1c | Diabetic nephropathy |
| C3, C4 | ↓C3+C4 in MPGN (classic pathway), ↓C3 only in C3 GN/DDD |
Footnote - Recent Literature
A 2026 systematic review (PMID 41981519) examined IgM nephropathy as a distinct entity vs. part of the podocytopathy spectrum - reinforcing the evolving classification of podocyte-injury disorders that you'll encounter on biopsy. This is relevant when you see IgM staining in a case that otherwise looks like MCD or early FSGS.
Robbins & Kumar Basic Pathology, Ch. 12 | NKF Primer on Kidney Diseases 8e, Ch. 16 | Brenner & Rector's The Kidney, Ch. 31 | Frameworks for Internal Medicine, Ch. 34
This is a shared conversation. Sign in to Orris to start your own chat.