Give me answer on Cystic fibrosis mention pathogenesis in flowchart for md exams 10 marks answer from fishman and murray and recent ats guidelines
Reading File
Finding Sources
Searching PubMed
"cystic fibrosis"[MeSH Terms] AND "management"[MeSH Terms]
Searching the Web
ATS cystic fibrosis guidelines 2024 2025
Reading File
Finding Sources
Searching PubMed
"cystic fibrosis" AND "CFTR modulator"
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive textbook data from both Murray & Nadel and Fishman's. I also have the actual pathophysiology flowchart diagram from Murray & Nadel. Now let me compile the complete MD exam answer.
CYSTIC FIBROSIS - MD Exam Answer (10 Marks)
Sources: Murray & Nadel's Textbook of Respiratory Medicine (Chapter 67), Fishman's Pulmonary Diseases and Disorders (Chapter 48), Recent Evidence (2024-2025)
DEFINITION
Cystic Fibrosis (CF) is the most common monogenic lethal disease in white populations, caused by mutations in the CFTR gene (chromosome 7q31.2), encoding a 190-kDa ATP-binding cassette (ABC) transporter family anion channel. It affects ~1 in 2,500-3,500 live births in white populations and 1 in 17,000 in African Americans.
- Murray & Nadel, Chapter 67
GENETICS
| Feature | Detail |
|---|---|
| Inheritance | Autosomal recessive |
| Gene | CFTR (chromosome 7q) |
| Mutations | >2000 variants; 350 confirmed disease-causing |
| Most common | F508del (p.Phe508del) - present in ~85% of patients |
| Carrier rate | 1 in 25 whites (~4%) |
CFTR Mutation Classes (Murray & Nadel):
| Class | Defect | Example | Treatment Target |
|---|---|---|---|
| I | No protein synthesis (nonsense/stop) | G542X | PTC suppressors |
| II | Misfolding/trafficking defect | F508del | Correctors (e.g., tezacaftor, elexacaftor) |
| III | Gating defect (protein reaches surface, won't open) | G551D | Potentiators (ivacaftor) |
| IV | Reduced conductance | R117H | Potentiators |
| V | Reduced synthesis | 3849+10kbC>T | Amplifiers |
| VI | Reduced stability at surface | - | Stabilizers |
PATHOGENESIS FLOWCHART
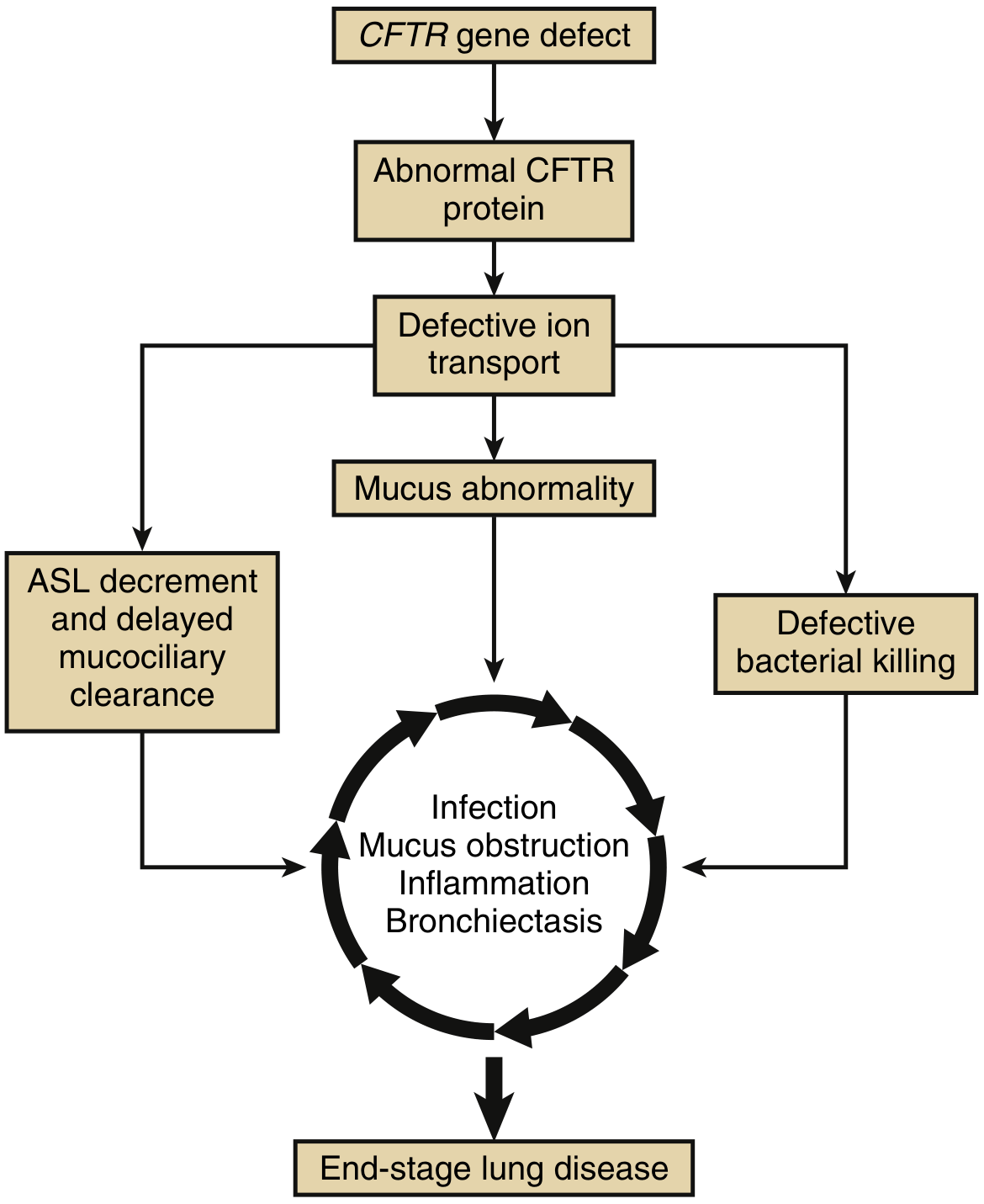
Figure 67.4 from Murray & Nadel - CF lung disease pathophysiology cycle
CFTR Gene Mutation (Chromosome 7q31.2)
|
▼
Defective/Absent CFTR Protein
(ABC Transporter, 27 exons, 2 MSDs, 2 NBDs, R domain)
|
▼
Defective Anion Transport
(↓ Cl⁻ secretion + ↑ Na⁺ absorption via ENaC)
(↓ HCO₃⁻ secretion)
/ \
▼ ▼
Dehydrated ASL Impaired Mucin Unfolding
(Airway Surface (↓ HCO₃⁻ → acidic milieu →
Liquid depleted) mucins don't expand on exit
from submucosal glands)
\ /
▼ ▼
MUCUS ABNORMALITY
(↑ MUC5B + MUC5AC expression driven by IL-1β)
(+ DNA from necrotic neutrophils → ↑ viscosity)
(Viscosity ∝ [mucin]³)
|
┌────┴──────────────────────┐
▼ ▼
ASL Decrement Defective Bacterial
+ Delayed Mucociliary Killing
Clearance (↓ thiocyanate → ↓
(Cilia compressed, lactoperoxidase activity)
motility impaired) (↓ antimicrobial peptides)
\ /
└──────────┬──────────────┘
▼
┌────────── VICIOUS CYCLE ──────────────┐
│ │
│ Bacterial Colonization │
│ (S. aureus early → P. aeruginosa │
│ mucoid late → B. cepacia complex) │
│ ↓ │
│ Neutrophil recruitment │
│ (IL-8, IL-1β, TNF-α) │
│ ↓ │
│ Exuberant Neutrophilic Inflammation │
│ (proteases, ROS, DNA release) │
│ ↓ │
│ Airway Wall Destruction │
│ + Bronchiectasis │
│ ↓ │
│ ↑ Mucus obstruction │
│ → more infection → ... │
└────────────────────────────────────────┘
▼
END-STAGE LUNG DISEASE
(Respiratory failure, Cor pulmonale)
Based on Murray & Nadel Figure 67.4 and Fishman's Chapter 48
CFTR PROTEIN - STRUCTURE & FUNCTION
- Located on apical surface of epithelial cells and on novel ionocytes (a minority airway cell type discovered via single-cell sequencing - major site of CFTR expression)
- CFTR regulates Cl⁻ and HCO₃⁻ secretion, membrane potential, and indirectly modulates ENaC (Na⁺ channel)
- Acts as a PDZ-domain anchored membrane complex, regulating numerous apical ion transporters
- Murray & Nadel, Chapter 67
DETAILED LUNG PATHOGENESIS (Fishman's Chapter 48)
Step 1 - Ion Transport Failure
- CFTR mutations → imbalance between ENaC-mediated Na⁺ absorption and CFTR-mediated Cl⁻ secretion
- Deficient HCO₃⁻ secretion → impaired mucin unfolding as mucus exits gland ducts
- Deficient thiocyanate secretion → reduced lactoperoxidase-derived isothiocyanate (antimicrobial)
Step 2 - Mucus Dysfunction
- Concentrated mucus layer draws water from periciliary liquid → cilia compressed → mucociliary clearance fails
- Biophysical properties scale with third power of mucin concentration (modest increase = dramatic viscosity increase)
- Increased MUC5B + MUC5AC (driven by IL-1β)
- DNA from necrotic neutrophils further increases mucus rigidity
Step 3 - Infection (Characteristic Organisms)
| Age/Stage | Organism |
|---|---|
| Early childhood | Haemophilus influenzae, Staphylococcus aureus |
| Later childhood | Pseudomonas aeruginosa (non-mucoid) |
| Advanced disease | P. aeruginosa (mucoid phenotype - biofilm former) |
| Terminal/severe | Burkholderia cepacia complex (worst prognosis) |
| Emerging | Stenotrophomonas maltophilia, NTM (Mycobacterium abscessus) |
Step 4 - Inflammation
- Disproportionate neutrophilic inflammation (even before detectable infection in newborn pigs)
- IL-8, IL-1β, TNF-α, LTB4 dominate
- Neutrophil elastase, MMP, ROS destroy airway walls
- Protease-antiprotease imbalance
Step 5 - Structural Lung Disease
- Small airway obstruction → tree-in-bud opacities (early HRCT)
- Progressive bronchiectasis (upper lobe predominant)
- Hyperinflation, air trapping
- Eventually: respiratory failure, pulmonary hypertension, cor pulmonale
EXTRAPULMONARY MANIFESTATIONS
| System | Manifestation | Mechanism |
|---|---|---|
| Pancreas (exocrine) | Steatorrhoea, fat-soluble vitamin deficiency | Duct obstruction → autodigestion |
| Pancreas (endocrine) | CF-related diabetes (CFRD) | Islet destruction |
| GI tract | Meconium ileus (neonates), DIOS in adults, rectal prolapse | Thick secretions |
| Liver | Focal biliary cirrhosis, multilobar cirrhosis | Bile duct plugging |
| Sinuses | Pansinusitis, nasal polyps | Same ion transport defect |
| Reproductive | Congenital bilateral absence of vas deferens (CBAVD) - 98% males infertile | Agenesis/obstruction |
| Sweat glands | High sweat [Cl⁻] >60 mEq/L | CFTR absent in sweat duct → Cl⁻ not reabsorbed |
| Bone | Osteopenia/osteoporosis | Malnutrition, chronic inflammation |
DIAGNOSIS
- Newborn screening - immunoreactive trypsinogen (IRT)
- Sweat chloride test (gold standard):
-
60 mEq/L = positive (CF)
- 30-59 mEq/L = intermediate (borderline)
- <30 mEq/L = normal
-
- CFTR mutation analysis (genetic testing - 2-allele identification)
- Nasal potential difference (CFTR function when sweat test equivocal)
- Clinical features: chronic sinopulmonary disease, exocrine pancreatic insufficiency, male infertility
MANAGEMENT (Fishman's + Murray & Nadel + CFF Guidelines 2024)
A. CFTR Modulator Therapy (Disease-Modifying)
| Drug | Mechanism | Indication |
|---|---|---|
| Ivacaftor (Kalydeco) | Potentiator - opens CFTR gate | Class III (G551D + 22 other gating mutations) |
| Lumacaftor/Ivacaftor (Orkambi) | Corrector + potentiator | F508del homozygous |
| Tezacaftor/Ivacaftor (Symdeko) | Corrector + potentiator | F508del homozygous or heterozygous |
| Elexacaftor/Tezacaftor/Ivacaftor (Trikafta) | Dual corrector + potentiator | ≥1 F508del allele (~90% of CF patients) |
Triple therapy (ETI/Trikafta) improves FEV1 by ~14%, decreases exacerbations by ~63%, and dramatically improves quality of life. It is now the standard of care for eligible patients. (Murray & Nadel, Chapter 68; Fishman's Chapter 48)
Recent 2024 data (PMID 39117676 - Mall et al., Nat Rev Dis Primers): ETI has shifted the disease trajectory but does not eliminate lung disease; patients with advanced disease prior to ETI availability will still require lung transplantation.
B. Airway Clearance
- Hypertonic saline (7%) - hydrates ASL, restores mucociliary clearance
- Dornase alfa (DNase) - cleaves extracellular DNA in mucus, reduces viscosity
- Chest physiotherapy / oscillating PEP devices (Flutter, Acapella)
- Inhaled mannitol - osmotic agent
C. Anti-infective Therapy
- Inhaled tobramycin (TOBI) - for chronic P. aeruginosa (28-day on/off cycle)
- Inhaled aztreonam - alternative for P. aeruginosa
- Inhaled colistin - for resistant gram-negatives
- Azithromycin - chronic anti-inflammatory use (reduces exacerbations via immunomodulatory effect)
- IV antibiotics for acute exacerbations (anti-pseudomonal combinations)
D. Nutritional Support
- High-calorie diet (120-150% of normal caloric requirement)
- Pancreatic enzyme replacement therapy (PERT)
- Fat-soluble vitamins (A, D, E, K)
- Salt supplementation (especially in hot weather)
E. Lung Transplantation
Indications (Fishman's/ISHLT criteria):
- FEV1 <25% predicted
- FEV1 <50% with 20% relative decline over 12 months
- Hypoxemia (PaO2 <60 mmHg) / Hypercapnia (PaCO2 >50 mmHg)
- Frequent hospitalizations, massive hemoptysis, rapid lung function decline
- CFF recommends early referral (before FEV1 <50%) to optimize medical/psychosocial status
PROGNOSIS
- With ETI modulators, median predicted survival has improved dramatically (>50 years in current era)
- Pre-ETI era: median survival ~37 years
- Major causes of death: respiratory failure (~80%), lung transplant complications
- B. cepacia complex colonization: significantly worsened transplant outcomes
SUMMARY TABLE FOR EXAM
| Feature | Detail |
|---|---|
| Gene | CFTR, chromosome 7q31.2 |
| Protein | 180-kDa ABC transporter, Cl⁻/HCO₃⁻ channel |
| Most common mutation | F508del (Class II - misfolding) |
| Key pathogenesis | ASL depletion → mucus stasis → chronic infection → neutrophilic inflammation → bronchiectasis |
| Key organism | Mucoid P. aeruginosa (chronic); B. cepacia (worst) |
| Diagnostic gold standard | Sweat chloride >60 mEq/L |
| Disease-modifying Rx | ETI (Trikafta) - for ≥1 F508del allele |
| Mucolytic | Dornase alfa (DNase) |
| ASL rehydration | Hypertonic saline |
Key References:
- Murray & Nadel's Textbook of Respiratory Medicine, Chapter 67 (CFTR and CF pathogenesis) and Chapter 68 (CFTR modulator therapy)
- Fishman's Pulmonary Diseases and Disorders, Chapters 6 and 48
- Mall MA et al. "Cystic fibrosis." Nat Rev Dis Primers. 2024 Aug 8. [PMID: 39117676]
- Terlizzi V & Lopes-Pacheco M. "Cystic fibrosis: new challenges and perspectives beyond elexacaftor/tezacaftor/ivacaftor." Ther Adv Respir Dis. 2025. [PMID: 40163448]
- Savant AP. "Cystic Fibrosis Year in Review 2024." Pediatr Pulmonol. 2025. [PMID: 40778614]
This is a shared conversation. Sign in to Orris to start your own chat.