Patbogenesis of hep c virus
Reading File
Finding Sources
Finding Sources
Searching PubMed
"hepatitis C virus" AND pathogenesis
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive, authoritative information from Harrison's 22nd Edition and Sherris & Ryan's Medical Microbiology 8th Edition. I can now compose a detailed, well-structured answer.
Pathogenesis of Hepatitis C Virus (HCV)
1. The Virus - Key Biology
HCV is a positive-sense, single-stranded RNA virus (family Flaviviridae, genus Hepacivirus). It exists as a collection of quasispecies - closely related but genetically distinct variants within a single host, generated by error-prone viral RNA replication. The replication rate is extraordinarily high (~1 × 10¹² virions/day), with approximately 10% of hepatocytes infected at any time. This high replication and genetic diversity is the cornerstone of its ability to evade both innate and adaptive immunity.
2. Entry and Cellular Tropism
HCV is transmitted via blood/blood products and initially infects peripheral blood B and T lymphocytes and monocytes, then homes to its primary site - the liver. Entry into hepatocytes involves multiple cell-surface receptors, including CD81, scavenger receptor class B type I (SR-BI), claudin-1, and occludin. These receptors, along with host factors, explain HCV's strict liver tropism.
3. Innate Immune Evasion - How HCV "Blinds" the First Line of Defense
After HCV enters a hepatocyte, the host identifies viral product motifs via pattern recognition receptors (PRRs) - particularly TLR3, RIG-I, and MDA5 - that distinguish viral dsRNA intermediates from "self." This triggers interferon (IFN) production and activation of innate immune responses.
However, HCV has evolved multiple strategies to disrupt this at every step:
| HCV Protein | Mechanism of Innate Immune Evasion |
|---|---|
| NS3/4A protease | Cleaves TRIF and MAVS (CARDIF), blocking type I interferon signaling |
| Core protein | Interacts with TNF receptor, decreasing cytolytic T-cell activity |
| NS4B, NS5B | Suppress the NF-κB immunoregulatory pathway, reducing antiapoptotic proteins and increasing vulnerability to TNF-α-mediated cell death |
| Envelope glycoproteins E1/E2 | High mutation rate creates quasispecies diversity, allowing escape from antibody neutralization |
The net effect is blocking of type I interferon responses and inhibition of interferon signaling effector molecules.
- Harrison's Principles of Internal Medicine 22E, p. 2692-2693
- Sherris & Ryan's Medical Microbiology, 8th Ed., p. 519
4. NK Cell Response (Innate Arm)
Natural killer (NK) cells respond to HCV-infected hepatocytes by:
- Releasing perforins that fragment nuclei of infected cells and induce apoptosis
- Secreting IFN-γ, which recruits intrahepatic inflammatory cells, stimulates the Th1 response, and induces necrosis/apoptosis of HCV-infected hepatocytes
In patients who spontaneously clear HCV, NK cells show enhanced IFN-γ production and upregulated activating receptors. In persistent infection, both peripheral and intrahepatic NK cytotoxicity become dysfunctional - an important reason for chronic infection.
5. Adaptive Immune Response and Liver Injury
CD8+ Cytotoxic T Lymphocytes (CTLs)
HCV itself is not directly cytopathic to hepatocytes under ordinary circumstances. Liver injury is primarily immune-mediated. Intrahepatic HLA class I-restricted CD8+ CTLs, directed at nucleocapsid and other viral antigens, are the primary effectors of hepatocyte killing. They eliminate infected cells via:
- Apoptosis of infected hepatocytes
- IFN-γ-induced inhibition of viral replication
Critically, the CTL response is less effective in chronic HCV compared to acute infection - contributing to viral persistence. Both T-cell exhaustion and HCV-driven immune escape mutations of CTL epitopes contribute to this impaired response.
CD4+ Helper T Cells
CD4+ T cells play an important role by secreting proinflammatory cytokines related to hepatocyte death. The balance between Th1 and Th2 cytokines is central to disease progression:
| Cytokine Type | Cytokines | Clinical Effect |
|---|---|---|
| Th1 | IL-2, TNF-α, IFN-γ | Aggressive hepatic disease, hepatocyte injury |
| Th2 | IL-10 | Milder presentation, promotes viral persistence |
TNF-α causes hepatic injury and triggers a "cytokine storm", causing liver damage in chronically infected patients.
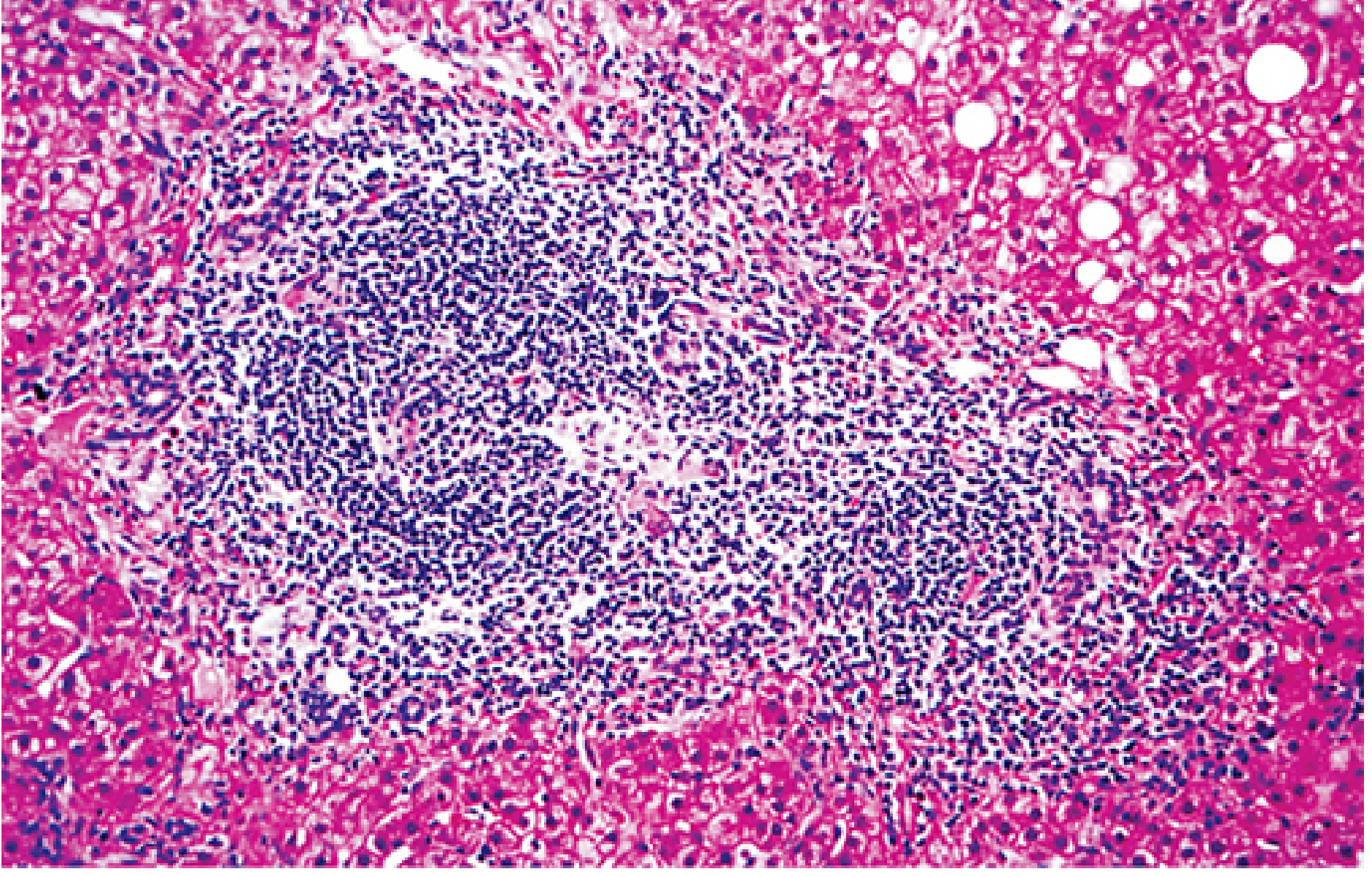
Histology of chronic HCV infection showing chronic portal inflammation with a prominent lymphoid aggregate (center), and piecemeal necrosis at the interface between portal connective tissue and parenchyma - the hallmark of chronic active hepatitis. (Sherris & Ryan's Medical Microbiology, 8th Ed.)
6. Humoral Immune Response and Immune Complex-Mediated Damage
Anti-HCV antibodies appear several weeks after infection (IgM and IgG), directed against core, envelope (E1/E2), NS3, and NS4 proteins. However:
- HCV antibodies are relatively low in titer and do not neutralize effectively
- Selective pressure from humoral immunity drives mutations in E1/E2 envelope proteins, allowing immune escape and persistent infection
- Antibodies form immune complexes that deposit in tissues and cause extrahepatic manifestations
Extrahepatic Immune Complex Damage:
- Vasculitis
- Arthritis
- Glomerulonephritis (membranoproliferative pattern)
- Cryoglobulinemia (mixed, type II) - the most characteristic extrahepatic manifestation
- Autoantibodies (antinuclear antibodies, anti-cytochrome P450 antibodies, liver-kidney microsomal antibodies)
7. Host Genetic Factors
Genetic host factors substantially influence HCV pathogenesis:
- IL28B gene (chromosome 19) - codes for interferon-λ3. The CC haplotype of IL28B is strongly linked with spontaneous HCV clearance. Non-CC polymorphisms (CT or TT) are associated with failure to clear infection. This has been mechanistically explained by a frameshift variant (ΔG polymorphism) creating an ORF in a novel interferon gene IFN-λ4, associated with impaired HCV clearance.
- MHC class II DR5 allele - associated with a lower incidence of cirrhosis in HCV-infected individuals
- HLA-A2 - CTLs restricted by HLA-A2 have been found in 97% of chronic hepatitis C patients
Extrinsic factors accelerating progression: alcohol abuse, smoking, co-infection with HIV/HBV/HAV, older age, and male sex.
8. Mechanism of Chronicity (~75-85% of patients)
The transition to chronic infection is the defining feature of HCV and results from multiple overlapping mechanisms:
- High viral mutation rate generating quasispecies diversity that outruns immune containment
- T-cell exhaustion - HCV-specific CD8+ CTLs become functionally exhausted during persistent antigen exposure (upregulation of PD-1, CTLA-4 inhibitory receptors)
- Impaired CD4+ T-cell help - leading to poor CD8+ responses
- Dysfunctional NK cells - particularly in patients with unfavorable IL28B alleles
- Antibody escape via envelope protein mutations
- Suppression of IFN signaling by viral non-structural proteins
9. Fibrosis and Hepatocellular Carcinoma (HCC)
Chronic necro-inflammation drives activation of hepatic stellate cells → progressive hepatic fibrosis → cirrhosis. From cirrhosis, risk of HCC is approximately 1-5% per year.
HCV promotes hepatocarcinogenesis via both indirect (inflammation-driven regeneration) and direct mechanisms:
- HCV core protein perturbs the cell cycle, interacts with components of growth regulatory pathways, upregulates oncogenes, and causes loss of tumor suppressor gene function
- NS3 and NS5A have also been implicated in oncogenic signaling
- Chronic HCV promotes insulin resistance in hepatocytes through TNF-α and IL-6 upregulation and oxidative stress, which itself drives fibrosis progression
Summary Diagram of Pathogenesis
HCV entry (via CD81, SR-BI, claudin-1)
↓
Viral replication (~10¹² virions/day)
↓
Pattern recognition (RIG-I, TLR3) → IFN production
↓
HCV evades (NS3/4A, core, NS4B, NS5B block IFN signaling)
↓
├── NK cells → perforin/IFN-γ → some viral control
└── Adaptive immunity
├── CD8+ CTLs → apoptosis of hepatocytes (main injury mechanism)
├── CD4+ T cells → TNF-α, IL-2 → cytokine-mediated injury
└── Antibodies → immune complexes → extrahepatic disease
Quasispecies diversity + T-cell exhaustion + impaired NK function
↓
CHRONICITY (75-85%)
↓
Necro-inflammation → stellate cell activation → Fibrosis → Cirrhosis → HCC
Sources:
- Harrison's Principles of Internal Medicine, 22nd Ed. - Pathogenesis chapter, pp. 2692-2693
- Sherris & Ryan's Medical Microbiology, 8th Ed., pp. 519-521
- Jawetz, Melnick & Adelberg's Medical Microbiology, 28th Ed., p. 525
This is a shared conversation. Sign in to Orris to start your own chat.