Opioid Various Mechanisms of Action
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a thorough, textbook-grounded breakdown of the various mechanisms by which opioids act.
Opioid Mechanisms of Action
1. Receptor Classes
Opioids act at four G protein-coupled receptors (GPCRs):
| Receptor | Gene | Primary Endogenous Ligands | Key Locations |
|---|---|---|---|
| Mu (μ / MOR) | OPRM1 (chr 6) | β-endorphin, enkephalins | Neocortex, caudate-putamen, nucleus accumbens, VTA, PAG, medulla, dorsal horn |
| Kappa (κ / KOR) | OPRK1 (chr 8) | Dynorphins | Caudate-putamen, hypothalamus, pituitary, amygdala, dorsal horn |
| Delta (δ / DOR) | OPRD1 (chr 1) | Enkephalins | Olfactory cortex, neocortex, nucleus accumbens, amygdala |
| Nociceptin / OFQ (NOPr) | OPRL1 (chr 20) | Nociceptin / N/OFQ | Most widely distributed; modulates pain, reward, anxiety, memory |
All three classical receptors share ~65% amino acid sequence homology and signal through Gi/o heterotrimeric G proteins. NOPr shows ~48-49% homology and is classified separately - notably, it is not blocked by naloxone.
- Goodman & Gilman's Pharmacological Basis of Therapeutics, p. 463-464
- Katzung's Basic and Clinical Pharmacology 16e, p. 874-875
2. Core Intracellular Signaling Cascade
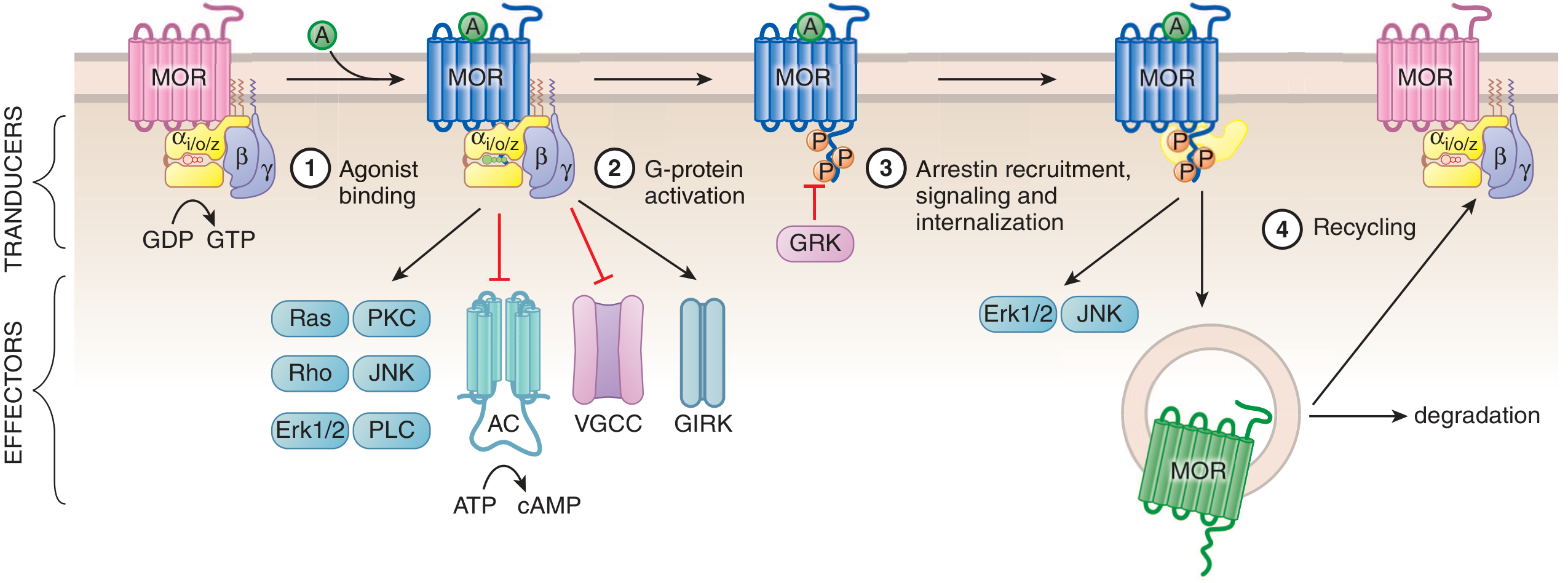
Figure: Simplified scheme of opioid receptor (MOR) signaling showing G-protein activation (Steps 1-2), arrestin-mediated internalization (Step 3), and receptor recycling or degradation (Step 4). - Goodman & Gilman's
When an opioid agonist binds, the receptor converts from its inactive (Ri) to active (Ra) conformation. This causes the α subunit of the Gi/o heterotrimer to exchange GDP for GTP, releasing the βγ dimer. Both subunits act on downstream effectors:
α-Subunit Effects
- Inhibits adenylyl cyclase (AC) → reduces cAMP → reduces PKA activity → decreased phosphorylation of many proteins and reduced cAMP-dependent Ca²⁺ influx
- On chronic opioid exposure, AC becomes supersensitized; abrupt withdrawal causes a "cAMP overshoot" - a key molecular basis of withdrawal symptoms
βγ Dimer Effects
-
Closes voltage-gated Ca²⁺ channels (VGCCs) on presynaptic terminals → reduced Ca²⁺ influx → inhibited neurotransmitter release
-
Opens GIRK channels (G protein-coupled inwardly rectifying K⁺ channels) → K⁺ efflux → membrane hyperpolarization → decreased neuronal firing
-
Activates MAPK/ERK and JNK pathways (important for long-term neuroadaptations)
-
Activates Ras, Rho, PKC, PLC signaling cascades
-
Goodman & Gilman's, p. 465
-
Katzung 16e, p. 874
3. Pre- and Postsynaptic Actions
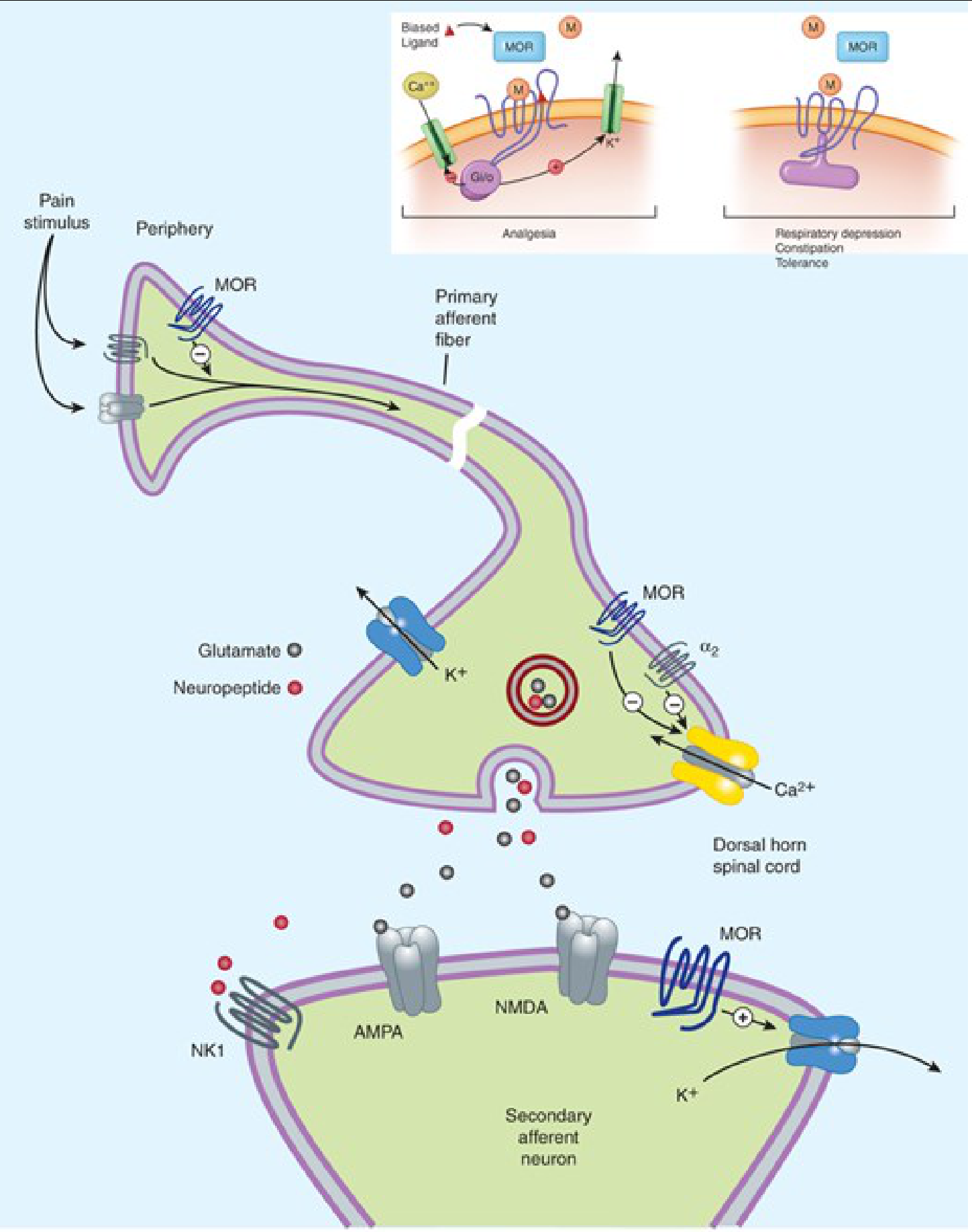
Figure: MOR-mediated analgesia at presynaptic terminals (closing VGCCs, inhibiting transmitter release) and postsynaptic neurons (opening K⁺ channels, hyperpolarizing). Inset shows how biased agonism at MOR leads to either analgesia (G-protein path) or adverse effects via arrestin path. - Katzung 16e
Presynaptic action (dominant site for analgesia):
- VGCCs close → reduced release of glutamate (main excitatory amino acid from nociceptive terminals), substance P, acetylcholine, norepinephrine, and serotonin
- This attenuates transmission of pain signals from primary to secondary afferent neurons in the dorsal horn
Postsynaptic action:
- K⁺ channel opening → hyperpolarization of secondary neurons → reduced action potential generation
Peripheral action:
-
Functional μ receptors exist on peripheral sensory nerve endings
-
Particularly relevant in inflammation; immune cells in injured tissue release β-endorphin, activating peripheral MOR
-
This reduces sensory neuron activity and transmitter release locally
-
Katzung 16e, p. 874-875
4. Receptor Subtypes and Their Physiological Effects
| Effect | Receptor Involved |
|---|---|
| Analgesia (supraspinal & spinal) | μ, δ, κ |
| Euphoria / reward | μ (via dopamine release in nucleus accumbens from VTA) |
| Respiratory depression | μ |
| Physical dependence | μ |
| Sedation | μ, κ |
| Dysphoria / psychotomimesis | κ |
| Miosis | μ, κ |
| Decreased GI motility | μ (enteric nervous system) |
| Diuresis regulation | κ |
| Mood modulation | δ |
The μ receptor was originally defined by its pharmacological profile using the relative analgesic potencies of opioid alkaloids. Analgesia, euphoria, respiratory depression, and physical dependence all result principally from μ receptor activation.
- Katzung 16e, p. 875
5. Descending Pain Modulation
Opioids activate descending antinociceptive pathways via disinhibition:
-
In the ventrolateral periaqueductal gray (PAG), opioids inhibit GABA interneurons → releases inhibition on descending pain-modulating neurons (raphe nucleus, noradrenergic nuclei) → these project to the dorsal horn to further suppress pain transmission
-
In the ventral tegmental area (VTA), inhibition of GABA interneurons → increased dopamine release in the nucleus accumbens → underlies reward and addictive potential
-
Goodman & Gilman's, p. 465
6. Receptor Structure
Opioid receptors are Class A GPCRs with:
-
Extracellular N-terminus
-
7 transmembrane (TM) alpha-helical domains connected by 3 extracellular and 3 intracellular loops
-
Intracellular C-terminus (palmitoylated, forming an extra small alpha-helix)
-
Orthosteric binding site within the TM domains - the nitrogen atom and phenolic OH group of morphine-like molecules (or the Tyr moiety of endogenous peptides) form a salt bridge with a conserved aspartate in TM3 and a water-mediated interaction with a histidine in TM6
-
Larger endogenous peptides (e.g., dynorphin) extend beyond the orthosteric pocket and interact with extracellular loops for receptor selectivity
-
Goodman & Gilman's, p. 465
7. Signal Termination: Desensitization and Internalization
- GRK (GPCR receptor kinase) phosphorylates the activated, agonist-bound receptor at intracellular loop and C-terminus residues
- β-arrestin is recruited to the phosphorylated receptor → sterically uncouples the receptor from G proteins (desensitization)
- β-arrestin scaffolds ERK1/2 and JNK signaling (arrestin-biased signaling)
- The receptor-arrestin complex is internalized into endosomes
- Internalized receptor is either recycled to the plasma membrane (resensitization) or targeted for lysosomal degradation (downregulation)
This is the molecular basis of acute tolerance to opioids.
- Goodman & Gilman's, p. 465 (Figure 23-2)
8. Biased Agonism (Functional Selectivity)
A critical concept in modern opioid pharmacology: different ligands stabilize different receptor conformations, producing differential activation of G-protein vs. arrestin signaling pathways from the same receptor.
- G-protein biased agonists preferentially activate the Gi/o pathway (analgesia, euphoria) while minimizing β-arrestin recruitment (reducing tolerance, constipation, and possibly respiratory depression)
- Oliceridine (approved IV opioid) was designed as a G-protein biased MOR agonist, though clinical evidence shows its safety profile is similar to morphine
- Mice with targeted deletion of β-arrestin develop less morphine tolerance - but still develop dependence - suggesting biased agonism has complex trade-offs
The endogenous enkephalins themselves represent a "balanced" ligand at MOR and may be a better therapeutic model for avoiding opioid dependence than biased synthetic drugs.
- Kaplan & Sadock's Comprehensive Textbook of Psychiatry, p. 590
- Goodman & Gilman's, p. 465
9. Partial Agonists and Antagonists
-
Partial agonists (e.g., buprenorphine) bind the orthosteric site but cannot produce the same maximal response as full agonists regardless of dose; buprenorphine's very high μ-receptor affinity means it displaces full agonists and requires much higher doses of naloxone for reversal
-
Antagonists (e.g., naloxone, naltrexone) bind with high affinity but produce no receptor activation; naloxone has ~10x higher affinity for μ than κ and reverses respiratory depression within 2 minutes IV; naltrexone has longer oral duration (up to 24 hours per dose)
-
Allosteric modulators act at a distinct non-orthosteric site: positive allosteric modulators can amplify endogenous opioid signaling without directly activating the receptor - a potential future strategy to reduce side effects
-
Lippincott Pharmacology, p. 727-728
-
Goodman & Gilman's, p. 465
Summary Diagram of Key Mechanisms
Opioid binds MOR/KOR/DOR
↓
Gi/o activation
↙ ↘
α subunit βγ dimer
↓ ↙ ↘
↓ Adenylyl Close VGCC Open GIRK (K⁺)
cyclase (presynaptic) (postsynaptic)
↓ cAMP ↓ ↓
↓ PKA ↓ Transmitter Hyperpolarization
release ↓ Firing
(Glutamate, SP,
NE, 5-HT, ACh)
↓
ANALGESIA (+ euphoria, sedation, respiratory depression)
Also via βγ: MAPK/ERK → long-term neuroplasticity, addiction
Sources: Goodman & Gilman's Pharmacological Basis of Therapeutics (23rd chapter); Katzung's Basic & Clinical Pharmacology 16e (Chapter 31); Lippincott Illustrated Reviews Pharmacology; Kaplan & Sadock's Comprehensive Textbook of Psychiatry
Other Mechanisms
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Loading Image
Reading File
I now have comprehensive source material. Here is the full answer on opioid "other mechanisms":
Opioids: Other Mechanisms of Action
Beyond the primary Gi/o-GPCR cascade covered previously, opioids exert actions through several distinct additional mechanisms - each clinically significant.
1. Monoamine Reuptake Inhibition (Tramadol & Tapentadol)
Some opioid-class analgesics achieve their effect through a dual mechanism - combining weak μ-receptor agonism with monoamine reuptake inhibition:
| Drug | μ-Receptor Activity | Monoamine Reuptake Effect |
|---|---|---|
| Tramadol | Weak (prodrug - active M1 metabolite has 200x higher affinity) | Inhibits both serotonin (5-HT) AND norepinephrine reuptake |
| Tapentadol | Moderate direct agonist (no prodrug conversion needed) | Inhibits norepinephrine only (no serotonin effect) |
How it works: By blocking reuptake of norepinephrine and serotonin in the spinal cord and brain, these drugs enhance the activity of descending inhibitory pain pathways - the same pathways normally modulated by endogenous opioids. Norepinephrine activates α2 receptors in the dorsal horn to suppress pain transmission; serotonin (via 5-HT) further inhibits nociceptive signaling.
Clinical implications:
-
Tramadol's serotonin effect creates risk of serotonin syndrome (especially with SSRIs/MAOIs)
-
Tramadol is a prodrug dependent on CYP2D6 for conversion to active M1; poor metabolizers (8% of Whites, up to 7% of African Americans, <0.5% of Asians) have attenuated analgesia
-
Tapentadol avoids CYP2D6 dependency (glucuronidation instead) - lower seizure risk and no serotonin syndrome risk compared to tramadol
-
Barash Clinical Anesthesia 9e, p. 4678; Katzung 16e, p. 1027
2. NMDA Receptor Interaction and Opioid-Induced Hyperalgesia (OIH)
With chronic opioid exposure, a counteradaptive mechanism involving NMDA receptors emerges:
Mechanism:
- Chronic MOR occupancy activates PKC (protein kinase C)
- PKC phosphorylates and upregulates/sensitizes NMDA glutamate receptors in spinal dorsal horn neurons
- Enhanced NMDA receptor activity increases excitability of pain-transmission neurons (central sensitization)
- The net result is paradoxical increased pain sensitivity - opioid-induced hyperalgesia (OIH)
OIH is:
- A paradoxical phenomenon where pain sensitivity increases during or after escalating opioid treatment
- Particularly well-documented after remifentanil infusions during anesthesia
- Distinct from tolerance (tolerance = reduced effect; OIH = opposite effect - more pain)
Therapeutic relevance:
-
Ketamine (an NMDA antagonist) can prevent or reverse OIH - hence its co-administration with opioids in perioperative settings
-
Methadone uniquely among opioids has intrinsic NMDA antagonist activity, which may explain its utility in neuropathic pain and lower propensity for tolerance
-
Goodman & Gilman's, p. 474 (system-level counteradaptation); Miller's Anesthesia 10e, p. 2893
3. Adenylyl Cyclase Superactivation - Molecular Basis of Withdrawal
With long-term opioid use, AC undergoes adaptive counterregulation:
- Normally, opioids inhibit AC → lower cAMP
- Chronically, cells compensate by upregulating AC expression and activity (superactivation)
- When opioid is suddenly withdrawn → the upregulated, now-uninhibited AC produces a massive cAMP overshoot
- This "cAMP storm" drives the autonomic surge of withdrawal: tachycardia, hypertension, diarrhea, hyperthermia, agitation, hyperalgesia, mydriasis, hormonal surges (ACTH, cortisol, pituitary hormones)
This AC superactivation represents cellular-level physical dependence and is mechanistically distinct from psychological craving/addiction.
- Goodman & Gilman's, p. 474
4. Reward and Mesocorticolimbic Dopamine Disinhibition
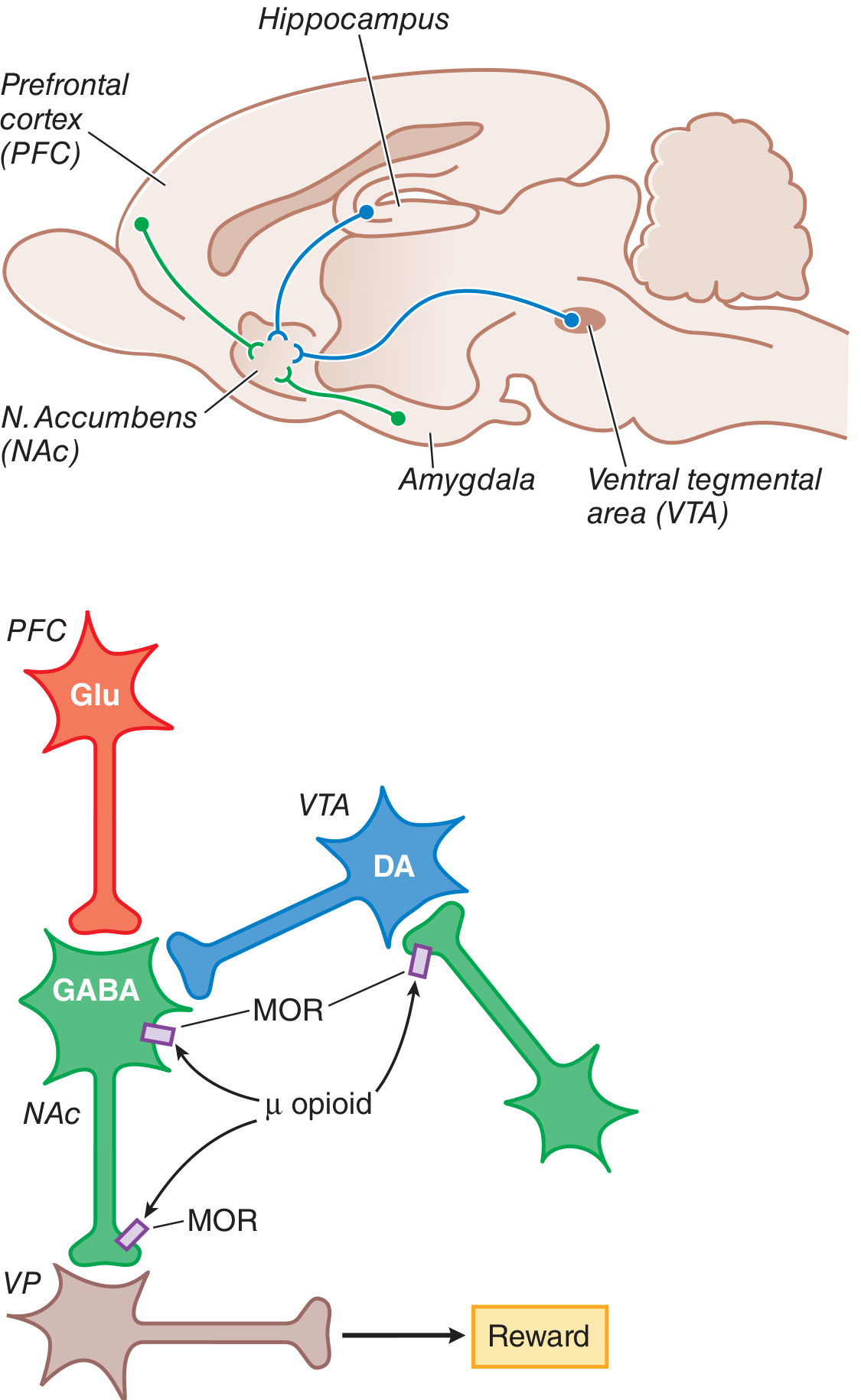
Figure: MOR agonists inhibit GABAergic interneurons in the VTA, releasing inhibition on dopamine neurons, which then increase DA release into the nucleus accumbens - the substrate of opioid reward. - Goodman & Gilman's
The rewarding properties of opioids operate through a disinhibition circuit, not direct dopamine receptor agonism:
- In the VTA, tonically active GABA interneurons normally suppress dopamine neuron firing
- MOR activation inhibits these GABA interneurons (by closing VGCCs and opening GIRK channels)
- With GABA inhibition removed, dopamine neurons fire more freely → increased DA release into the nucleus accumbens (NAc)
- DA in NAc projects to the ventral pallidum (VP) → generates the positive reinforcing (reward) state
- MOR on NAc neurons also directly reduces GABA output to VP, further enhancing the reward signal
This mesocorticolimbic circuit (VTA → NAc → VP, with inputs from PFC, hippocampus, amygdala) is the shared reward pathway for opioids, alcohol, and other drugs of abuse.
- Goodman & Gilman's, p. 474-475
5. System-Level Tolerance: Differential Tolerance Development
Not all opioid effects develop tolerance at the same rate - a mechanistically important observation:
| Effect | Tolerance Develops? |
|---|---|
| Analgesia | Yes - relatively rapidly |
| Sedation | Yes |
| Euphoria | Yes |
| Respiratory depression | Yes (but dangerous window remains) |
| Constipation (GI motility) | Minimal - persists with chronic use |
| Miosis | No - persists even in highly tolerant patients (diagnostic sign of opioid toxicity) |
Why? Miosis requires activation of only a small fraction of the receptor pool (low fractional occupancy requirement). Analgesic and sedative effects require larger fractional activation, which is more susceptible to receptor downregulation and uncoupling. Additionally, receptor homo- and heterodimer formation may alter trafficking and contribute to differential tolerance.
- Goodman & Gilman's, p. 474
6. Non-Neuronal Mechanisms: Immune System
Opioids also act through non-neuronal pathways:
- Opioid receptors are expressed on macrophages, peripheral microglia, and astrocytes
- Delta-opioid receptors (DOR) on cardiac tissue may afford cardioprotection during ischemia
- β-endorphin released from immune cells in inflamed tissue activates peripheral MOR to produce local analgesia - particularly relevant in inflammatory pain states
- This peripheral immune-mediated opioid effect is why intra-articular opioids (e.g., post-arthroscopy) can provide up to 24 hours of analgesia
These immune-opioid interactions are increasingly recognized as relevant in inflammatory pain management and may eventually yield peripherally selective opioid compounds with fewer CNS-mediated side effects (respiratory depression, addiction).
- Barash Clinical Anesthesia 9e, p. 1522; Goodman & Gilman's, p. 464
7. Endogenous Opioid Pathway Activation (Non-Drug Mechanisms)
Endogenous opioid circuits can be activated by:
- Stress-induced analgesia - extreme stress triggers endogenous opioid release
- Placebo-induced analgesia - naloxone-reversible, confirming genuine MOR mediation
- Conditioned pain modulation (CPM) - pain at one site is reduced by a second painful stimulus elsewhere ("pain inhibits pain"), mediated partly through endogenous opioid release
These mechanisms share circuitry with exogenous opioid drugs and explain cross-tolerance and overlap in analgesic pathways.
- Barash Clinical Anesthesia 9e, p. 1522
Summary: Mechanisms Beyond Classical GPCR Signaling
| Mechanism | Drug/Context | Key Effect |
|---|---|---|
| Monoamine reuptake inhibition | Tramadol (NE+5-HT), Tapentadol (NE only) | Enhanced descending inhibition |
| NMDA receptor sensitization (PKC-mediated) | Chronic opioid use | OIH, tolerance |
| NMDA antagonism | Methadone | Anti-neuropathic, reduced tolerance |
| AC superactivation | Chronic use/withdrawal | cAMP overshoot → withdrawal syndrome |
| Dopamine disinhibition (VTA GABA) | All MOR agonists | Reward, addiction liability |
| Differential fractional occupancy | All chronic opioid use | Persistent miosis, variable tolerance |
| Peripheral immune opioid receptors | Inflammatory states | Local analgesia without CNS effects |
| Endogenous pathway activation | Placebo, stress, CPM | Naloxone-reversible analgesia |
Sources: Goodman & Gilman's Pharmacological Basis of Therapeutics (pp. 474-475); Katzung's Basic & Clinical Pharmacology 16e (p. 1027); Barash's Clinical Anesthesia 9e (pp. 1522, 4678); Miller's Anesthesia 10e (p. 2893)
This is a shared conversation. Sign in to Orris to start your own chat.