Formate of ind with study required Importance and study need to ind submission Classify and explain the ind University longer expand answer in detail
Reading File
Finding Sources
Searching the Web
IND Investigational New Drug application format classification FDA requirements
Finding Sources
Reading Web Page
IND format classification types importance study requirement submission
Reading File
Reading File
Searching the Web
IND application phases clinical trials study needed preclinical data CMC pharmacology toxicology
Loading Image
Reading Web Page
IND content format sections cover sheet protocol chemistry manufacturing clinical pharmacology
I now have comprehensive information from authoritative sources. Let me compile the full, detailed university-level answer.
Investigational New Drug (IND) Application - Complete University-Level Guide
1. DEFINITION AND OVERVIEW
An Investigational New Drug (IND) Application is a formal request submitted by a clinical study sponsor to the U.S. Food and Drug Administration (FDA) for authorization to administer an investigational drug or biological product to human subjects in clinical trials. It is governed under 21 CFR Part 312 of the Code of Federal Regulations.
As stated in Goodman & Gilman's Pharmacological Basis of Therapeutics: "Before the drug candidate can be administered to human subjects in a clinical trial, the sponsor must file an Investigational New Drug (IND) application, a request to the FDA for permission to use the drug for human research. The IND describes the rationale and preliminary evidence for efficacy in experimental systems, as well as pharmacology, toxicology, chemistry, manufacturing, and so forth. It also describes the plan (protocol) for investigating the drug in human subjects."
Authorization under IND must be secured prior to interstate shipment and administration of any new drug or biological product that is not yet the subject of an approved New Drug Application (NDA) or Biologics License Application (BLA).
2. WHY AN IND IS NEEDED - IMPORTANCE AND STUDY REQUIREMENT
A. Legal Requirement
Under the Federal Food, Drug, and Cosmetic Act (FDCA), no new drug may be given to humans in clinical research without prior FDA authorization. An IND is the mechanism through which that authorization is obtained. Beginning human trials without a valid IND is a federal violation.
B. Human Subject Protection
The primary purpose of the IND process is to ensure that research subjects will not be subjected to unreasonable risk. The FDA has 30 calendar days after receipt to review an IND before clinical trials may begin. During this review window, the FDA can:
- Allow the study to proceed
- Place a clinical hold (partial or full) if safety concerns exist
- Request additional data
C. Preclinical to Clinical Bridge
All new drug candidates undergo two major phases before market approval:
- Preclinical (nonclinical) studies - animal-based assessment of safety, toxicology (acute, sub-acute, chronic), pharmacokinetics (ADME), pharmacodynamics, reproductive toxicity, mutagenicity, and carcinogenicity
- Clinical trials (Phases 1-4) - human-based evaluation of safety and efficacy
The IND serves as the official bridge between these two phases. Without satisfactory preclinical data compiled in an IND, no clinical testing in humans is permitted.
D. Regulatory Oversight Throughout Development
The IND is a living document that remains active throughout clinical development. It must be updated with:
- Protocol amendments (changes to study design)
- Information amendments (new CMC, pharmacology-toxicology, or clinical data)
- IND safety reports (15-day expedited reports for serious unexpected adverse reactions)
- Annual progress reports (filed within 60 days of the annual IND date)
E. Ethical Obligations
The IND framework integrates with Institutional Review Board (IRB) oversight, informed consent requirements (21 CFR Part 50), and financial disclosure rules (21 CFR Part 54) to protect trial participants.
3. CLASSIFICATION OF IND
A. By TYPE (Three IND Types)
| IND Type | Description |
|---|---|
| Commercial IND | Submitted by pharmaceutical or biotech companies intending to market the drug; subject to full regulatory requirements |
| Research (Non-commercial) IND | Submitted by academic investigators, government researchers, or non-profit organizations for research purposes without commercial intent |
| Emergency IND | Authorizes use of an experimental drug in emergency situations where there is no time to file a standard IND, or for patients who cannot access the drug through a protocol |
B. By CATEGORY (Two IND Categories)
| Category | Description |
|---|---|
| Investigator IND | Submitted by an individual investigator who both initiates and conducts the study, including direct supervision of drug use (sponsor-investigator model) |
| Sponsor IND | Submitted by a company, institution, or organization that takes responsibility for the study but does not personally conduct it; investigators are separately named |
C. Special IND Designations
| Designation | Description |
|---|---|
| Treatment IND | Allows access to experimental drugs for patients with serious or life-threatening conditions before final FDA review, after the drug has shown promise in early testing |
| Exploratory IND | Conducted very early in Phase 1 development with limited human exposure, below the therapeutic dose level, to gather PK/PD data with minimal safety risk (also called "Phase 0") |
| Expanded Access IND | Permits use of investigational drugs outside of clinical trials for patients with serious conditions who have no alternative treatments |
4. FORMAT OF THE IND APPLICATION (21 CFR § 312.23)
The IND must be submitted in the following order, as mandated by federal regulation. Commercial sponsors must use the electronic Common Technical Document (eCTD) format. Non-commercial sponsors are exempt from eCTD requirements but are encouraged to use it.
SECTION 1: Cover Sheet - FDA Form 1571
This is the mandatory cover document for every IND submission. It must contain:
- Full name, address, and telephone number of the sponsor
- Date of application
- Name of the investigational new drug
- Identification of the phase(s) of clinical investigation to be conducted (Phase 1, 2, or 3)
- A commitment that the sponsor will not begin clinical investigations until the IND is in effect
- A commitment to conduct investigations in accordance with all applicable regulations
- Certification that the sponsor will notify the FDA of any adverse experience reports
- Signature of the sponsor or sponsor's authorized representative
SECTION 2: Table of Contents
An organized listing of all sections and attachments in the IND for efficient FDA review navigation.
SECTION 3: Introductory Statement and General Investigational Plan
- Brief description of the drug substance and drug product
- The drug's route of administration, dosage level(s), and duration of administration
- The type of investigation(s) to be conducted
- The general plan for clinical investigation (including phases, goals, number of subjects, safety monitoring)
SECTION 4: Investigator's Brochure (IB)
A compilation of clinical and nonclinical data on the investigational drug relevant to the study of the product in human subjects. Must include:
- Physical, chemical, and pharmaceutical properties and formulation
- Pharmacological and toxicological data obtained from animal studies
- Summary of human experience (if any prior human data exists)
- Brief statement of potential risks and side effects based on all prior experience
- Any precautions or special monitoring required
SECTION 5: Clinical Protocol(s) - FDA Form 1572
The detailed study plan for each proposed investigation. Each protocol must include:
a. Study Objectives and Purpose
- Clear statement of the research question and hypothesis
b. Investigator Information
- Name, address, and credentials of each investigator (Form FDA-1572 - Statement of Investigator)
- Name, address of IRB responsible for review
c. Patient Selection Criteria
- Inclusion and exclusion criteria
- Age range, sex, and health status of subjects
- Number of subjects to be enrolled
d. Study Design
- Phase designation (I, II, or III)
- Randomization and blinding methods
- Comparator arm (placebo, standard of care)
e. Dose, Route, and Duration
- Dose levels to be administered
- Method of administration
- Duration of individual subject participation
f. Safety Monitoring Plan
- Clinical observations to be made
- Laboratory tests to be performed
- Criteria for stopping the study (dose-limiting toxicity, stopping rules)
g. Statistical Analysis Plan
- Procedures for tabulating and analyzing data
- Primary and secondary endpoints
SECTION 6: Chemistry, Manufacturing, and Controls (CMC)
This section demonstrates that the investigational drug can be consistently produced and controlled at the required quality. It includes:
a. Drug Substance Information
- Name, structural formula, and molecular weight
- Method of synthesis/manufacture
- Physical, chemical, and biological properties
- Stability data and storage conditions
- Purity, impurity profile, and analytical test methods
b. Drug Product Information
- Formulation composition (active ingredient, excipients)
- Dosage form and strength
- Container/closure system
- Manufacturing process and controls
- Release specifications and in-process controls
c. Placebo Formulation (if applicable)
d. Labeling
- All labels and labeling to be used for the investigational product
e. Environmental Analysis
- Assessment of potential environmental impact (most products qualify for categorical exclusion)
SECTION 7: Pharmacology and Toxicology Information
This section provides the scientific basis for believing the drug is reasonably safe for initial human testing. It is divided into:
a. Pharmacology (Animal)
- Mechanism of action
- Pharmacodynamic effects (receptor binding, downstream effects)
- Pharmacokinetics (ADME): absorption, distribution, metabolism, excretion
- Drug-drug interaction data
b. Toxicology Studies
- Acute toxicity: Single-dose studies in at least two species; establishes LD50 and initial safety margins
- Subacute/Subchronic toxicity: Repeated-dose studies (14 days to 3 months) assessing target organ toxicity
- Chronic toxicity: Required for drugs intended for long-term human use (6 months to 2 years)
- Genotoxicity/Mutagenicity: Ames test, chromosomal aberration assay, in vivo micronucleus test
- Reproductive and developmental toxicity: Effects on fertility, embryo-fetal development (teratogenicity), postnatal development
- Carcinogenicity: Required if long-term use is intended; rat and mouse lifetime studies
- Local tolerance: Assessment at the intended route of administration
The test results provide the Maximum Tolerated Dose (MTD), which guides the starting dose in Phase 1 human trials.
SECTION 8: Previous Human Experience (if any)
- Summary of all prior human use of the drug, whether from foreign studies, compassionate use, or unapproved marketed use
- Any published or unpublished results of prior human testing
- If the drug has been marketed elsewhere, information about those markets
SECTION 9: Additional Information
Any other information that would aid evaluation of the proposed clinical investigations, including:
- Drug dependence and abuse potential (if applicable)
- Radioactivity data (if radioactively labeled drug)
- Pediatric considerations (if the drug will be tested in children)
- Special manufacturing requirements
KEY FDA FORMS IN THE IND
| Form | Purpose |
|---|---|
| FDA Form 1571 | Cover sheet - IND application |
| FDA Form 1572 | Statement of Investigator - binds the investigator to protocol compliance |
| FDA Form 3674 | Certification of registration in ClinicalTrials.gov |
5. STUDY REQUIREMENTS CORRESPONDING TO IND PHASES
The IND framework corresponds directly to the four phases of clinical development:
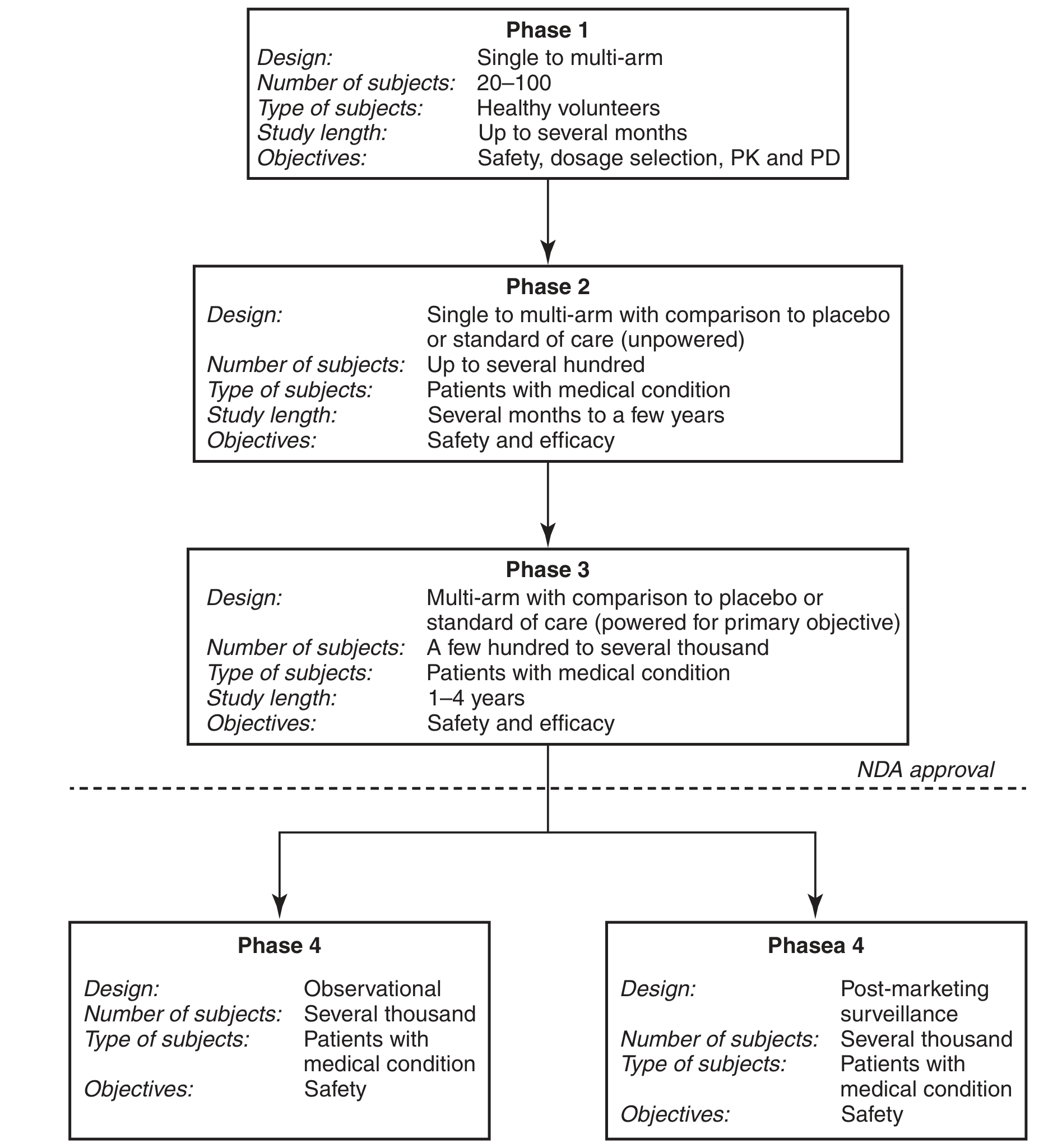
(Drug development clinical study phases - Tietz Textbook of Laboratory Medicine, 7th Ed.)
| Phase | Subjects | N | Duration | Primary Objectives |
|---|---|---|---|---|
| Phase 1 | Healthy volunteers | 20-100 | Up to several months | Safety, dose selection, PK/PD |
| Phase 2 | Patients (target disease) | Up to several hundred | Months to a few years | Safety and efficacy (preliminary) |
| Phase 3 | Patients (target disease) | Hundreds to thousands | 1-4 years | Definitive safety and efficacy |
| Phase 4 | Patients (post-market) | Several thousand | Ongoing | Long-term safety surveillance |
Phase 1 studies under the IND are the first human trials. Their intent is to "determine the metabolism and pharmacologic actions of the drug in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on efficacy" (Tietz Textbook of Laboratory Medicine).
After Phase 3 completion, the sponsor submits a New Drug Application (NDA) or Biologics License Application (BLA) for market approval. The IND technically remains active until the NDA/BLA is approved.
6. ONGOING IND OBLIGATIONS AFTER INITIAL SUBMISSION
Once an IND is active, the sponsor must maintain compliance through:
- 30-Day Review Window - No clinical trial begins until 30 days post-submission (or sooner if FDA clears it)
- Protocol Amendments - Required before implementing any protocol changes
- IND Safety Reports - Serious Unexpected Suspected Adverse Reactions (SUSARs) must be reported within 15 calendar days (7 days for fatal/life-threatening reactions)
- Annual Reports - Filed within 60 days of the IND anniversary, including enrollment progress, safety summary, protocol status, and manufacturing changes
- Information Amendments - New CMC, pharmacology/toxicology, or clinical data submitted as needed (but no more than every 30 days where feasible)
7. IND EXEMPTIONS
An IND is NOT required when:
- A lawfully marketed drug is being studied under conditions of approved labeling (routine clinical practice)
- The study does not involve a route of administration, dose level, or patient population that significantly increases risk
- The investigation does not involve a new indication or purpose that requires demonstrating safety and effectiveness
- The study is not intended to support an NDA or IND application
Notably, the FDA has determined that an IND is not currently required for Fecal Microbiota Transplantation (FMT) for treatment of refractory Clostridioides difficile infection under certain conditions (Yamada's Textbook of Gastroenterology).
8. SUMMARY DIAGRAM OF IND PROCESS
DISCOVERY & PRECLINICAL
↓
Synthesize molecule → Animal pharmacology/toxicology → CMC development
↓
PRE-IND MEETING WITH FDA (optional but recommended)
↓
SUBMIT IND APPLICATION (Form 1571 + all sections)
↓
FDA 30-DAY REVIEW
├── Approved → Begin Phase 1 Clinical Trial
├── Clinical Hold → Additional data required
└── Disapproved → Cannot proceed
↓
PHASES 1 → 2 → 3 (ongoing IND amendments/reports)
↓
NDA / BLA SUBMISSION → Market Approval
↓
PHASE 4 Post-Marketing Surveillance
9. KEY REGULATORY REFERENCES
| Regulation | Subject |
|---|---|
| 21 CFR Part 312 | IND Application (primary regulation) |
| 21 CFR Part 50 | Protection of Human Subjects / Informed Consent |
| 21 CFR Part 56 | Institutional Review Boards (IRBs) |
| 21 CFR Part 58 | Good Laboratory Practice (GLP) for nonclinical studies |
| 21 CFR Part 314 | NDA applications |
| 21 CFR Part 316 | Orphan Drug designations |
| 21 CFR Part 201 | Drug Labeling |
| 21 CFR Part 54 | Financial Disclosure by Clinical Investigators |
Sources:
- Goodman & Gilman's The Pharmacological Basis of Therapeutics (The IND Application section)
- Tietz Textbook of Laboratory Medicine, 7th Ed. (Drug Development, p. 385)
- U.S. Code of Federal Regulations, 21 CFR Part 312 (eCFR, 2025)
- FDA CBER: Submission of an IND to CBER
- PMC Article: Understanding FDA Regulatory Requirements for IND Applications - PMID 4435682
This is a shared conversation. Sign in to Orris to start your own chat.