Central pontine demyelination
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Here is a comprehensive overview of central pontine demyelination (osmotic demyelination syndrome):
Central Pontine Myelinolysis / Osmotic Demyelination Syndrome
The older term central pontine myelinolysis (CPM) has been replaced by the more accurate osmotic demyelination syndrome (ODS), since lesions are not always confined to the pons.
History & Epidemiology
The condition was first described by Adams and colleagues in 1959, based on four patients (mainly alcoholics) who developed rapidly evolving quadriplegia and pseudobulbar palsy. Postmortem examination revealed a large, symmetric demyelinative lesion in the base of the pons. The incidence is approximately 0.25% in consecutive adult autopsies. — Adams and Victor's Principles of Neurology, 12th Ed.
Pathology
The hallmark lesion is:
- Symmetric demyelination in the center of the base of the pons — may range from a few millimeters to involve almost the entire ventral pons
- There is always a rim of intact myelin between the lesion and the pons surface
- Microscopically: destruction of myelin sheaths with relative sparing of axons and pontine nuclear neurons
- Oligodendrocytes are depleted; reactive phagocytes and glial cells are present
- No inflammation — this distinguishes CPM from MS and postinfectious demyelination
- Because all damage occurs synchronously, all lesions appear at the same stage of myelin loss
The lesion may extend posteriorly into the medial lemnisci and tegmental structures but does not typically reach the medulla.
Extrapontine myelinolysis — identical lesions can occur in the internal capsule, deep cerebral white matter, corpus callosum, thalamus, subthalamic nucleus, striatum, amygdala, lateral geniculate body, and cerebellar folia.
— Robbins, Cotran & Kumar Pathologic Basis of Disease; Adams and Victor's Principles of Neurology
Etiology & Pathogenesis
The central event is a rapid rise in serum osmolality, most commonly from rapid correction of hyponatremia.
Key points:
- Initial serum sodium is typically <130 mEq/L (often much lower: 100–115 mEq/L)
- Experimental models (Laureno, 1983): dogs made severely hyponatremic then rapidly corrected with 3% saline developed spastic quadriparesis and identical pontine/extrapontine lesions
- In severely burned patients, extreme serum hyperosmolality alone — without antecedent hyponatremia — has also produced CPM
- ODS rarely occurs with serum [Na⁺] >120 mmol/L or with hyponatremia of <48 hours duration
Proposed mechanism: Hypotonicity downregulates sodium-coupled amino acid transporters (e.g., SNAT2), delaying return of osmolytes to the brain. When sodium is rapidly corrected, this delay causes cerebral dehydration and potential blood-brain barrier breakdown. Astrocytes appear to be early targets, activating microglia and proinflammatory cytokines. — Comprehensive Clinical Nephrology, 7th Ed.
High-risk conditions:
- Chronic alcoholism (most common — often co-exists with Wernicke disease)
- Chronic severe hyponatremia
- Liver failure / liver transplantation (incidence 13–29% at autopsy)
- Chronic renal failure on dialysis
- Malnutrition, cachexia, burns
- Lymphoma, advanced cancer
- Severe bacterial infections, dehydration, electrolyte disturbances
Clinical Features
Symptoms typically appear 2–6 days after rapid sodium correction (biphasic course):
- Phase 1: Generalized encephalopathy during rapid correction
- Phase 2 (2–3 days later): Behavioral changes, cranial nerve palsies, then progressive motor deterioration
Full syndrome:
- Flaccid quadriplegia (progressing to spasticity and extensor posturing)
- Pseudobulbar palsy — inability to chew, swallow, or speak
- Locked-in syndrome — fully conscious but unable to communicate or move, with intact comprehension and vertical eye movements
- Conjugate gaze limitations, nystagmus (in some cases)
- Pupils and facial sensation may be spared
Partial/mild syndrome:
- Confusion, dysarthria, cerebellar ataxia, disturbances of conjugate gaze — without quadriplegia
Many cases are clinically silent (especially small lesions), recognized only at autopsy or on MRI.
Imaging
MRI is the diagnostic modality of choice:
- T2-weighted / FLAIR: Symmetric hyperintensity in the central pons, characteristically involving transverse pontine fibers while sparing the descending corticospinal tracts (gives a "trident" shape)
- Diffusion restriction may appear in the lower pons within a day or so of motor signs; T2 changes typically appear several days later
- Non-enhancing lesions (no gadolinium enhancement)
- Periventricular and subpial regions are spared
- Imaging may be initially normal for up to 2 weeks — do not exclude the diagnosis based on early normal MRI
MRI (Harrison's 22E) — axial T2 through the pons showing symmetric pontine hyperintensity:
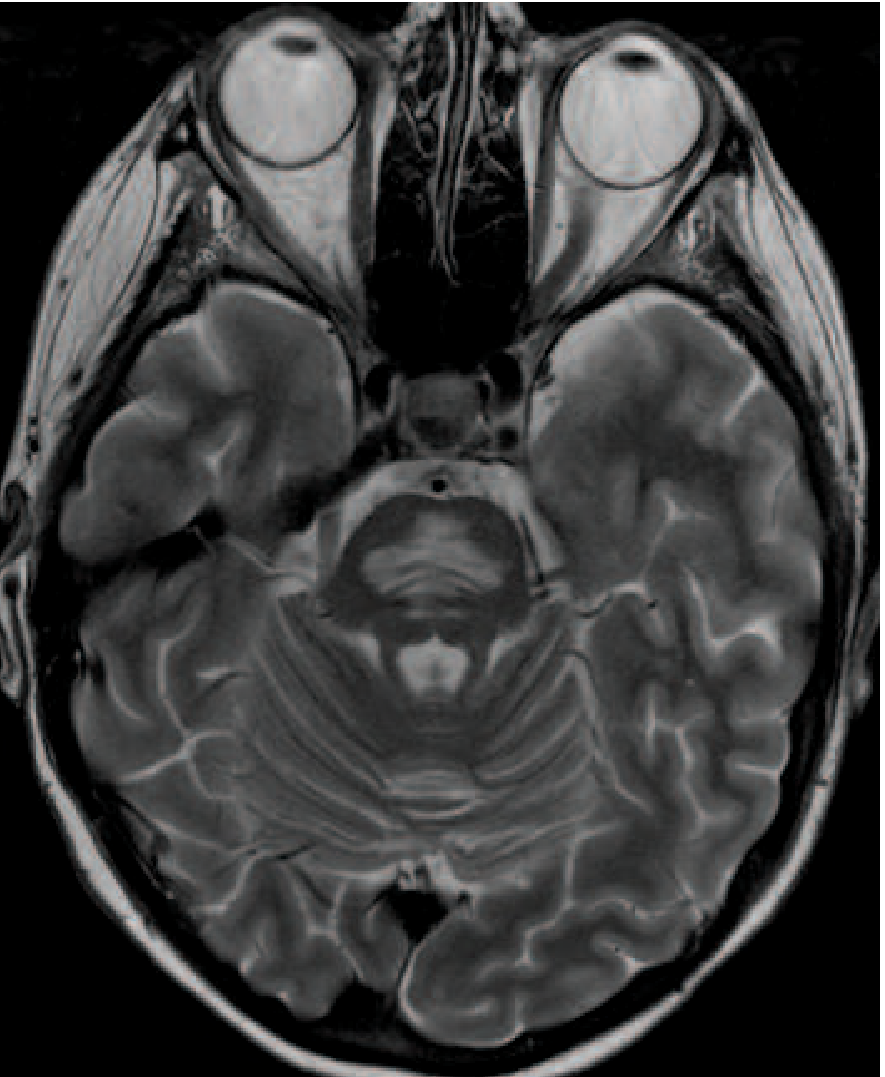
Axial T2 MRI showing symmetric pontine hyperintensity in ODS, with characteristic sparing of corticospinal tracts. — Harrison's Principles of Internal Medicine 22E, Fig. 318-5
Differentiating from brainstem infarction:
- Infarction: diffusion restriction on DWI, often extends to ventral surface of pons, asymmetric, may involve midbrain/thalamus
- ODS: T2 hyperintensity without restricted diffusion, spares ventral surface of pons
Prevention
The cornerstone of prevention is controlled, gradual correction of hyponatremia:
| Guideline | Rate |
|---|---|
| Harrison's 22E | ≤8 mmol/L per 24 h; ≤15 mmol/L per 48 h |
| Adams & Victor | No more than 6–8 mEq/L per 24 h |
| Karp & Laureno (classic) | ≤10 mEq/L in first 24 h; ≤21 mEq/L in first 48 h |
Prognosis
Originally considered uniformly fatal. However, a substantial proportion of patients show partial or even full neurologic recovery — even those with severe locked-in syndrome at onset — indicating that reversible forms exist. Some patients are left with permanent dysarthria, cerebellar ataxia, or other deficits. — Comprehensive Clinical Nephrology, 7th Ed.; Adams and Victor's Principles of Neurology
Sources: Harrison's Principles of Internal Medicine 22E | Adams and Victor's Principles of Neurology, 12th Ed. | Robbins, Cotran & Kumar Pathologic Basis of Disease | Comprehensive Clinical Nephrology, 7th Ed.
This is a shared conversation. Sign in to Orris to start your own chat.