Hpo axis?? And the evolution of female and male reproductive system
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Excellent - the images came through perfectly. Now I have everything needed for a thorough answer. Let me compile it all.
HPO Axis and Evolution of the Male & Female Reproductive Systems
Part 1: The HPO (Hypothalamic-Pituitary-Ovarian) Axis
The HPO axis is the master hormonal control system for female reproduction. It operates as a pulsatile feedback loop between three levels: the hypothalamus, the anterior pituitary, and the ovaries.
The Three Levels
1. Hypothalamus - GnRH
The hypothalamus releases Gonadotropin-Releasing Hormone (GnRH) in pulses, generally every 60 to 90 minutes. This pulsatility is absolutely required - a continuous (non-pulsatile) GnRH signal actually suppresses the axis (the basis of GnRH agonist therapy for conditions like endometriosis).
2. Anterior Pituitary - LH and FSH
GnRH pulses trigger the gonadotroph cells of the anterior pituitary to release:
- FSH (Follicle-Stimulating Hormone) - stimulates follicle growth and estradiol production by granulosa cells
- LH (Luteinizing Hormone) - drives theca cells to produce androgens and triggers ovulation
3. Ovaries - Estradiol, Progesterone, Inhibin
The ovaries respond to gonadotropins and feed back on the hypothalamus and pituitary.
The Menstrual Cycle: How the HPO Axis Runs It
The axis drives two parallel cycles: the ovarian cycle and the endometrial cycle.
Follicular / Proliferative Phase (Days 1-14)
- FSH and LH stimulate a cohort of ovarian follicles to grow
- All developing follicles produce estradiol, which gradually rises
- Rising estradiol stimulates rapid endometrial growth (proliferative phase)
- Early in this phase, estradiol exerts negative feedback - dampening GnRH/LH
- As estradiol rises further and stays elevated for >36-48 hours, it switches to positive feedback - a critical reversal that triggers the LH surge
The LH Surge and Ovulation
- The LH surge is an abrupt, dramatic rise around day 13-14, peaking ~12 hours after initiation and lasting ~48 hours
- Peak LH is roughly 3-fold above baseline
- The surge is triggered by persistently high estradiol, plus positive contributions from progesterone and activin
- The LH surge triggers ovulation: the dominant follicle ruptures and releases the oocyte
Luteal / Secretory Phase (Days 14-28)
- The ruptured follicle becomes the corpus luteum, which secretes both progesterone and estradiol
- Progesterone transforms the endometrium from proliferative to secretory (glandular phenotype) - preparing for implantation
- Progesterone + estradiol together exert strong negative feedback on GnRH/LH, suppressing new follicle development
- The corpus luteum also produces inhibin, which specifically suppresses FSH
- If fertilization does not occur, the corpus luteum degenerates (~day 26-28), progesterone/estradiol fall, negative feedback is removed, and menstruation begins - restarting the cycle
Key Feedback Summary
| Phase | Hormone | Feedback on HPO |
|---|---|---|
| Early follicular | Low estradiol | Negative feedback (low amplitude LH pulses) |
| Late follicular | High sustained estradiol | Positive feedback → LH surge |
| Luteal | Progesterone + estradiol | Negative feedback → suppresses new follicle growth |
| Luteal | Inhibin B | Suppresses FSH specifically |
Medical Physiology (Boron & Boulpaep), p. 1629 - "This feedback loop is unusual because it elicits negative feedback on the hypothalamic-pituitary axis throughout most of the menstrual cycle but positive feedback immediately before ovulation."
Part 2: Evolution (Development) of the Male and Female Reproductive Systems
Sexual differentiation is a stepwise process organized around three levels: genetic sex → gonadal sex → phenotypic sex.
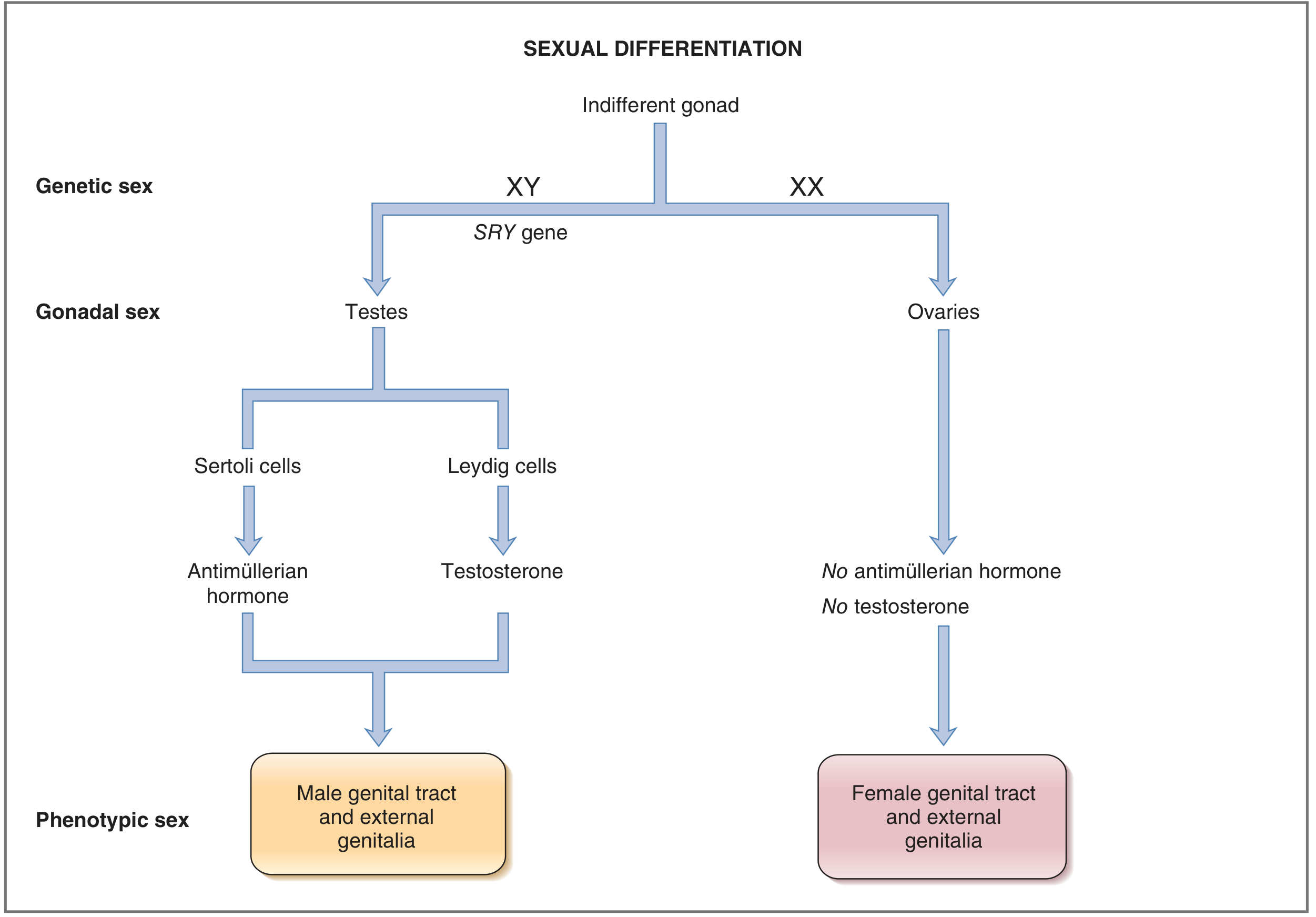
Step 1: Genetic Sex (Determined at Fertilization)
- XY = male; XX = female
- For the first ~5 weeks of gestation, gonads are indifferent (bipotential) - neither male nor female
- At ~week 7 in XY embryos, the SRY gene (sex-determining region of the Y chromosome) product activates, driving the indifferent gonad to develop into testes
- In XX embryos (no SRY), the gonads begin developing into ovaries slightly later (~week 9)
Step 2: Gonadal Sex
Testes (XY) contain three cell types:
- Germ cells → spermatogonia
- Sertoli cells → produce Anti-Mullerian Hormone (AMH), also called Mullerian-Inhibiting Substance (MIS)
- Leydig cells → produce testosterone
Ovaries (XX) contain three cell types:
- Germ cells → oogonia (arrested in meiosis I until ovulation)
- Granulosa cells → produce estradiol (with theca cells)
- Theca cells → produce progesterone and androgens (precursors for estradiol)
The key difference: the ovaries produce neither AMH nor testosterone. This turns out to be decisive.
Step 3: Phenotypic Sex - The Duct Systems
Every embryo starts with both duct systems in parallel:
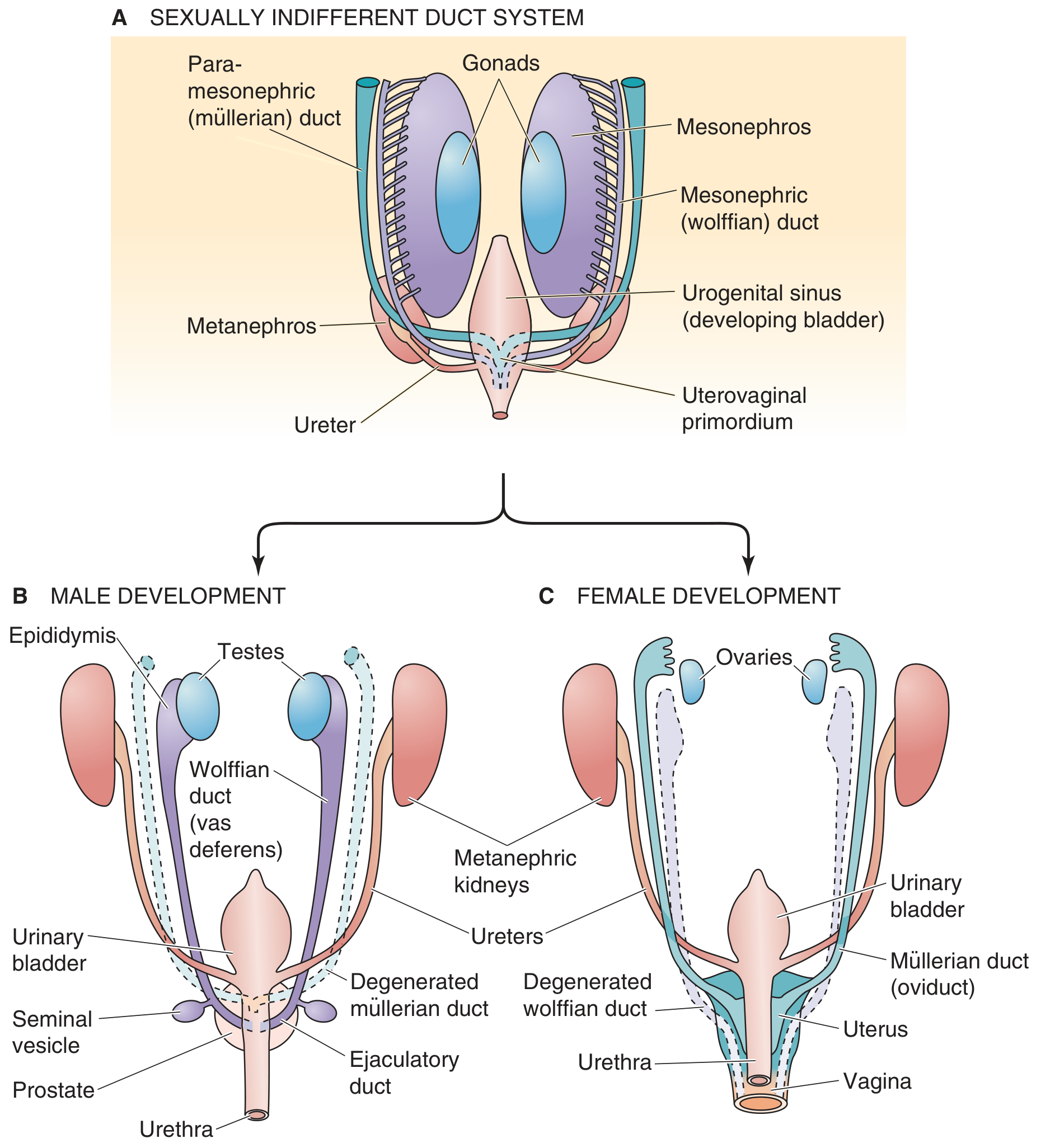
A. The Indifferent Stage (both sexes have both ducts):
- Wolffian (mesonephric) ducts - run from mesonephros to urogenital sinus
- Müllerian (paramesonephric) ducts - run parallel to wolffian ducts; fuse caudally to form the uterovaginal primordium
Male Development - Two Hormones Do the Work
AMH from Sertoli cells → causes regression and dissolution of the Müllerian ducts (which would otherwise become the uterus/tubes/upper vagina)
Testosterone from Leydig cells → stimulates survival and differentiation of the Wolffian ducts into:
- Epididymis (stores and matures sperm)
- Vas deferens (sperm transport)
- Seminal vesicles
- Ejaculatory duct
Additionally, testosterone is converted by 5α-reductase into dihydrotestosterone (DHT), which virilizes the external genitalia (see below).
If there are no testes, the fetus develops as female by default - no hormones are needed to make a female; female development is the "baseline" pathway.
Female Development - Default Pathway
- No AMH → Müllerian ducts survive and differentiate into:
- Fallopian tubes (oviducts)
- Uterus
- Cervix
- Upper 1/3 of the vagina (lower 2/3 from urogenital sinus)
- No testosterone → Wolffian ducts degenerate (small remnants may persist as Gartner's duct cysts)
Step 4: External Genitalia - Common Origin, Different Fates
Both sexes start with the same anlage:
| Embryonic Structure | Male (under DHT) | Female (no androgen) |
|---|---|---|
| Genital tubercle | Glans penis | Clitoris |
| Urogenital folds | Shaft of penis (fuse over urethra) | Labia minora (stay separate) |
| Labioscrotal swellings | Scrotum | Labia majora |
- In males, DHT (not testosterone itself) drives fusion of urogenital folds and enlargement of the phallus
- In females, the absence of androgens leaves the structures separate; the clitoris remains small
- External genitalia are anatomically indistinguishable until the end of the first trimester
Summary Table: Male vs. Female Differentiation
| Feature | Male (XY + SRY) | Female (XX, no SRY) |
|---|---|---|
| Gonad | Testes | Ovaries |
| Key hormones | AMH + Testosterone | None needed (default) |
| Müllerian ducts | Regress (AMH) | Persist → uterus, tubes, upper vagina |
| Wolffian ducts | Persist → epididymis, vas deferens, seminal vesicles | Regress |
| External genitalia | Penis, scrotum (DHT) | Clitoris, labia minora/majora |
Puberty: HPO Axis "Wakes Up"
Both systems converge at puberty, when the HPO axis (which was suppressed since infancy) becomes active again:
- Boys: GnRH → LH → Leydig cell proliferation → testosterone → spermarche, penile growth, voice change, facial hair, muscle mass
- Girls: GnRH → FSH/LH → estradiol from ovaries → breast budding (thelarche), then menarche ~2 years later; pubic/axillary hair (adrenarche) driven by adrenal androgens
Pulsatile GnRH is the key - intermittent pulses initiate puberty; continuous GnRH does not.
Sources: Costanzo Physiology 7th Ed., p. 461-469 | Boron & Boulpaep Medical Physiology, p. 1578-1582, 1621-1629
Pathology : rheumatic fever and rheumatic heart disease comprehensive
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Loading Image
Reading File
Loading Image
I now have comprehensive material from multiple authoritative sources. Let me compile the full answer.
Rheumatic Fever and Rheumatic Heart Disease - Comprehensive Pathology
1. Definition and Overview
Acute Rheumatic Fever (ARF) is an acute, immunologically mediated, multisystem inflammatory disease that occurs classically 2-3 weeks after Group A Streptococcal (GAS) pharyngitis (occasionally after GAS skin infection). It affects the joints, heart, skin, and CNS.
Rheumatic Heart Disease (RHD) is the long-term sequela of one or more episodes of ARF, characterized by deforming fibrotic valvular disease - most importantly mitral stenosis. RHD is virtually the only cause of acquired mitral stenosis.
Globally, ~33 million cases exist with ~300,000 annual deaths, with the highest burden in Oceania, South Asia, and central sub-Saharan Africa. It causes 25-45% of all cardiovascular disease in developing countries.
2. Epidemiology
- Peak age of ARF: 5-15 years (peak years for streptococcal pharyngitis)
- ARF occurs equally in males and females, but RHD is more common in women
- Peak prevalence of RHD: 3rd and 4th decades (cumulative valve damage from recurrent attacks)
- Lifetime cumulative incidence after rheumatogenic GAS: 3-6% regardless of geography, implying a fixed proportion of genetically susceptible individuals
- Genetic heritability of RF estimated at ~60%; family history increases risk ~5x
- HLA class II associations, cytokine gene polymorphisms (TNF-α, TGF-β), and B-cell alloantigens have been implicated but findings are inconsistent
3. Pathogenesis - Molecular Mimicry
The mechanism is molecular mimicry - the immune response to GAS cross-reacts with host cardiac tissues:
GAS Pharyngitis
↓ (2-3 week latent period)
Antibodies & CD4+ T cells against streptococcal M proteins
↓ (molecular mimicry)
Cross-react with cardiac antigens
(pericardial, myocardial, valvular proteins)
↓
Antibody-mediated + T cell-mediated cardiac injury
Specific mechanisms by organ:
| Manifestation | Immune Mechanism |
|---|---|
| Carditis | Cross-reactive antibodies + T-cell infiltration |
| Arthritis | Immune complex deposition in joints |
| Chorea | Antibodies bind basal ganglia / dopamine receptors → activate CaMKII in neurons |
| Skin (nodules, EM) | Delayed hypersensitivity |
Key molecular targets:
- N-acetyl-beta-d-glucosamine (GlcNAc) - group A carbohydrate epitope
- Laminin / laminar basement membrane in heart valve endothelium
- Cardiac myosin - cross-reactive with streptococcal M protein
- Collagen autoantibodies develop as collagen is released from damaged valves
The "two-hit" hypothesis: antibody attack on valve endothelium facilitates extravasation of T cells into valve tissue → formation of Aschoff bodies (granulomatous nodules).
Note: Streptococci are completely absent from the lesions - the damage is purely immunological.
4. Morphology (Pathologic Features)
4.1 Acute RF - The Aschoff Body (Pathognomonic)
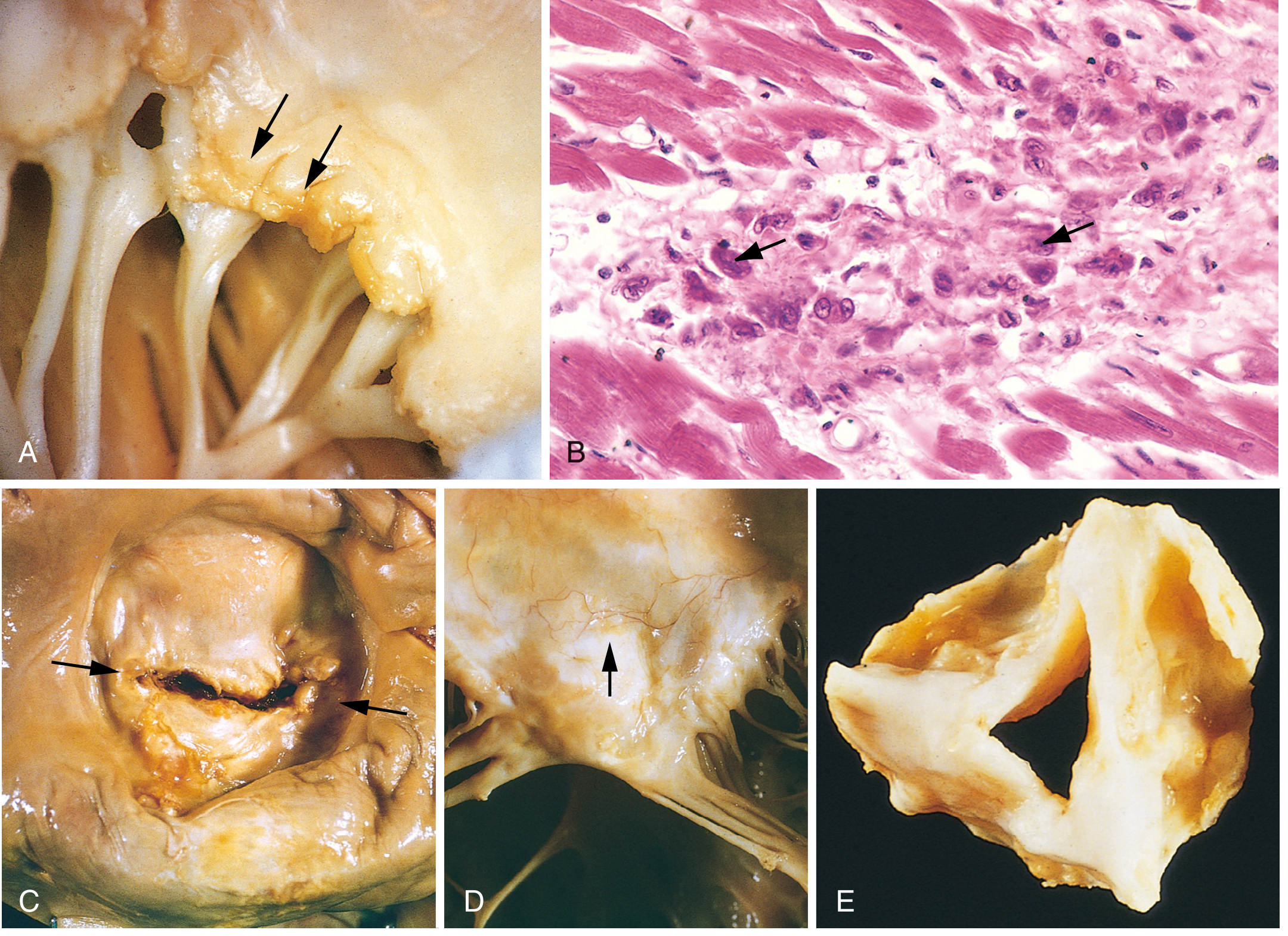
Fig. 12.22 - Robbins Pathology: (A) Acute rheumatic mitral valvulitis with small verrucae (arrows) along the line of closure. (B) Microscopic Aschoff body - the large cells with arrows are Anitschkow (caterpillar) cells. (C) Chronic RHD - "fish-mouth" mitral stenosis (arrows). (D) Thickened valve leaflet with neovascularization. (E) Severely fibrotic and fused aortic valve.
Aschoff bodies (also called Aschoff-Geipel bodies) are the hallmark lesion of RF myocarditis:
- Focal granulomatous inflammatory lesions
- Composed of: T lymphocytes + occasional plasma cells + Anitschkow cells
- Anitschkow cells (caterpillar cells): plump activated macrophages with abundant cytoplasm and central ovoid nucleus in which chromatin condenses into a slender, wavy ribbon resembling a caterpillar - pathognomonic of RF
- Aschoff bodies can be found in any of the three cardiac layers (pancarditis)
4.2 Acute RF - Pancarditis
Pericarditis: Fibrinous pericarditis, sometimes with effusion. Usually resolves without permanent damage.
Myocarditis: Diffuse inflammation with Aschoff bodies in myocardium. Rarely causes significant systolic dysfunction acutely.
Endocarditis (most clinically important):
- Inflammation of the endocardium and left-sided valves
- Fibrinoid necrosis within valve cusps or tendinous cords
- Small (1-2 mm) verrucae (vegetations) form along the lines of closure of affected valves
- These are distinct from the large vegetations of infective endocarditis
- MacCallum plaques: irregular subendocardial thickenings in the left atrium, caused by regurgitant jets
4.3 Valve Involvement Pattern
| Valve | Frequency |
|---|---|
| Mitral (alone) | ~65-70% |
| Mitral + Aortic | ~25% |
| Tricuspid | Infrequent |
| Pulmonary | Rarely |
4.4 Chronic RHD - The Healed Lesion
Chronic fibrotic changes are the consequence of healing after repeated acute attacks:
Cardinal anatomic changes of mitral valve in chronic RHD:
- Leaflet thickening (fibrosis)
- Commissural fusion - leads to stenosis; classically produces the "fish-mouth" or "buttonhole" appearance
- Shortening of leaflets
- Thickening and fusion of chordae tendineae
The fibrotic mitral valve obstructs left atrial outflow → mitral stenosis → left atrial enlargement → atrial fibrillation → risk of left atrial thrombus and systemic embolism.
5. Clinical Features of Acute RF
ARF typically develops 2-4 weeks after GAS pharyngitis. Note: up to 1/3 of patients develop RF after asymptomatic GAS infection.
Major Manifestations (Frequency)
| Feature | Frequency | Notes |
|---|---|---|
| Fever | >90% | Usually >38.5°C |
| Migratory polyarthritis | ~75% | Large joints (knees, ankles, elbows, wrists); "migrates" from joint to joint; very painful; sterile synovial fluid with lymphocyte predominance; does NOT cause permanent joint damage |
| Carditis | >50% | Most serious - pancarditis; valvulitis especially of mitral and aortic valves; initially causes regurgitation, not stenosis |
| Sydenham Chorea | ~30% | Involuntary, non-rhythmic, purposeless movements; limbs, face, trunk; more pronounced one side; disappears during sleep; caused by basal ganglia antibodies |
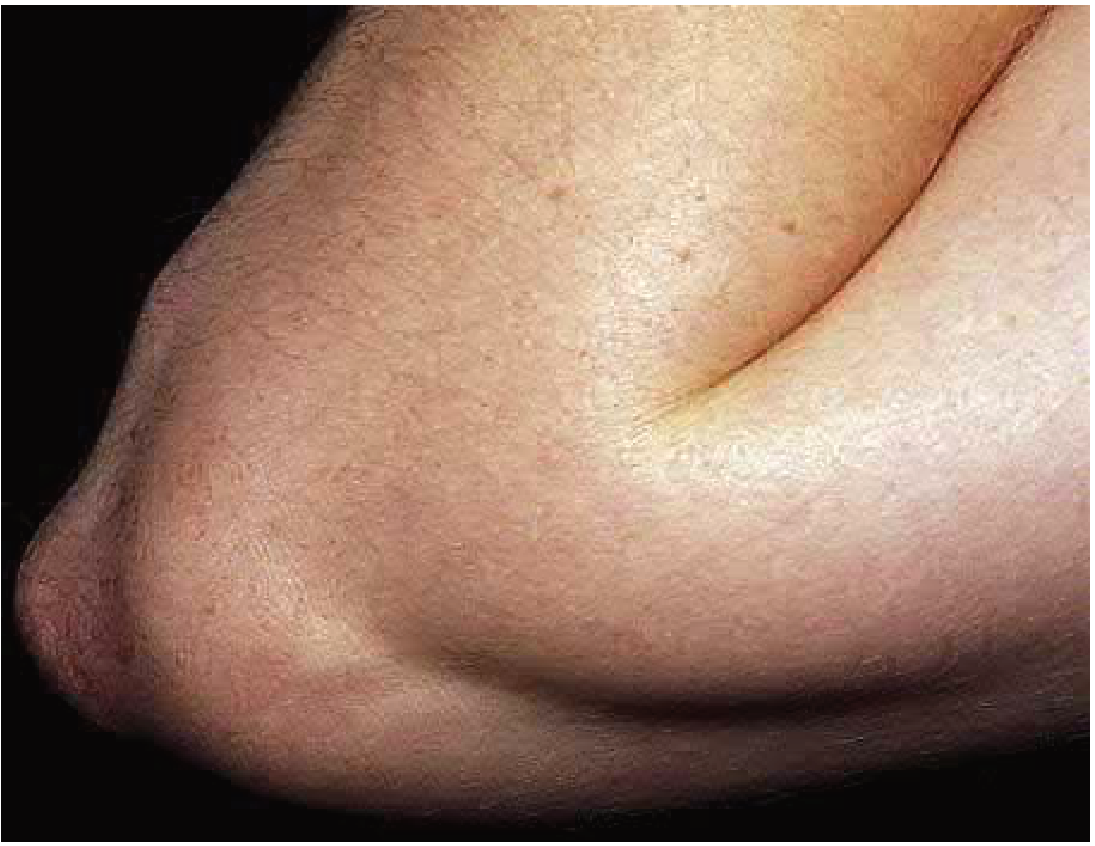
| Subcutaneous nodules | <10% | Firm, painless, freely movable; 0.5-2 cm; over bony prominences (elbows, wrists, knees, occiput); related to severity of carditis |
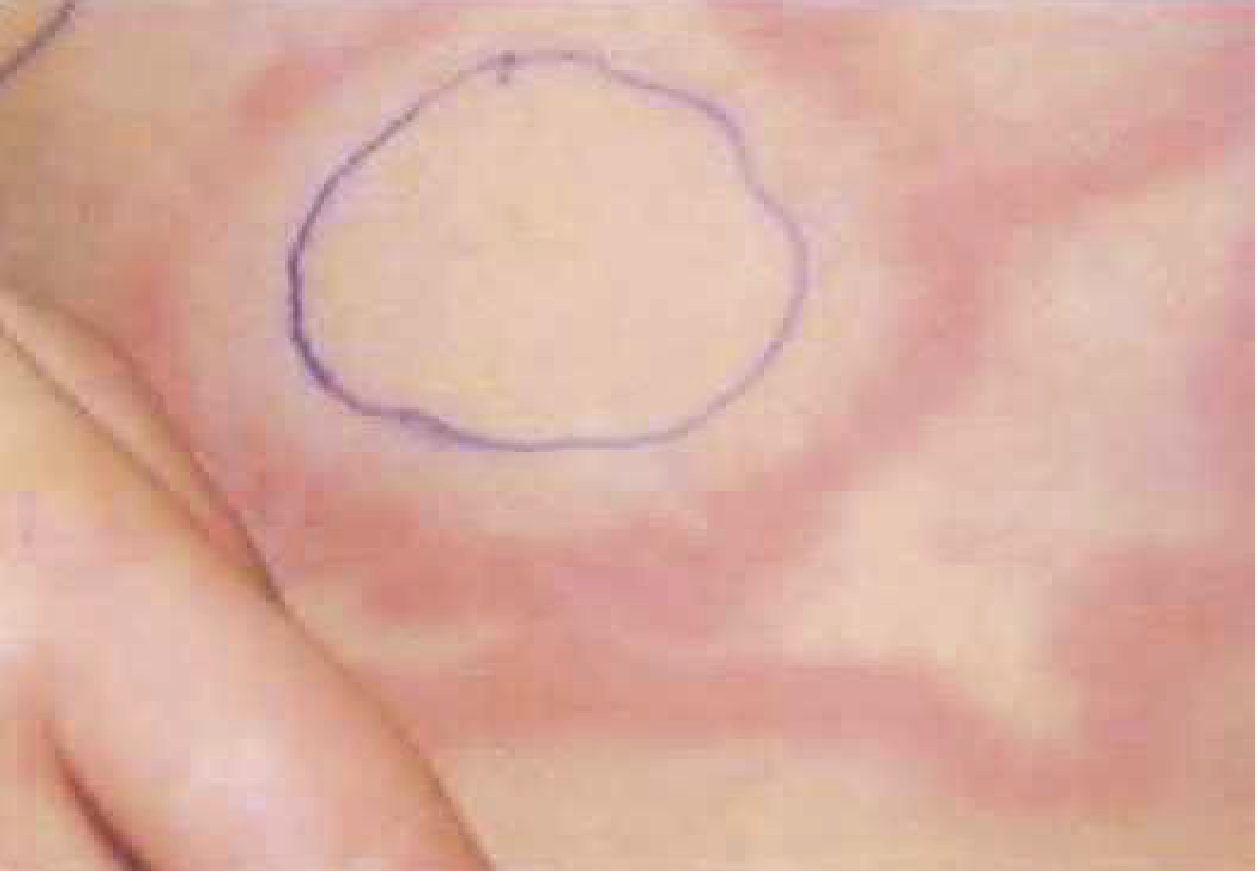
| Erythema marginatum | <10% | Serpiginous, flat/slightly raised, non-scarring, painless rash; trunk and proximal limbs; spreads outward leaving clear center; evanescent (moves hour to hour) |
Subcutaneous nodules (bony prominences):
Erythema marginatum (characteristic serpiginous rash):
6. Diagnosis - Revised Jones Criteria (2015, AHA)
ARF is a diagnosis of exclusion - no definitive single test exists.
Prerequisites (Evidence of Preceding GAS Infection)
- Positive throat culture or rapid strep antigen test
- Elevated or rising streptococcal antibody titer (ASO, anti-DNase B)
2015 Revised Jones Criteria
| Low-Risk Populations (ARF <2/100,000/yr) | Moderate/High-Risk Populations | |
|---|---|---|
| Diagnosis | 2 Major OR 1 Major + 2 Minor | 2 Major OR 1 Major + 2 Minor OR 3 Minor |
| Major criteria | Carditis (clinical/subclinical echo), Polyarthritis, Chorea, Erythema marginatum, Subcutaneous nodules | Carditis, Mono or Polyarthritis, Chorea, Erythema marginatum, Subcutaneous nodules |
| Minor criteria | Polyarthralgia, Fever ≥38.5°C, ESR ≥60mm and/or CRP ≥3.0 mg/dL, Prolonged PR interval | Monoarthralgia, Fever ≥38.5°C, ESR ≥30mm and/or CRP ≥3.0 mg/dL, Prolonged PR interval |
Key 2015 changes:
- Subclinical valvulitis on echocardiography accepted as major criterion in all populations
- Stratification into low-risk vs. moderate/high-risk populations
- Monoarthritis and polyarthralgia added as major criteria in high-risk populations
Supporting Investigations
- ECG: Prolonged PR interval in only 35% of cases; higher-degree block is uncommon
- Echocardiogram: Recommended for ALL patients with confirmed/suspected ARF - detects subclinical carditis even without murmurs; monitor chronic RHD
- Labs: Elevated ESR/CRP; normochromic normocytic anemia; leukocytosis; elevated ASO titer
7. Natural History and Complications
- Patients with RF have ~50% risk of recurrence with subsequent untreated GAS pharyngitis
- Joint inflammation resolves fully (no residual damage), typically within 1 month without treatment
- Murmurs disappear in ~50% of patients with mild acute carditis without major cardiac enlargement
- Chronic valvular disease can develop even in patients who appeared to recover fully from the acute episode
- Sydenham chorea: Usually resolves in months; ~1/3 have recurrences
- Prognosis depends primarily on:
- Severity of initial cardiac involvement
- Whether episodes are recurrent (each attack causes cumulative valve damage)
8. Management
A. Acute Attack Treatment
1. Eradicate GAS - Antibiotic Treatment:
- Single IM injection benzathine penicillin G (drug of choice) OR
- 10-day course of oral penicillin/amoxicillin OR
- Erythromycin (if penicillin-allergic)
2. Anti-inflammatory Therapy:
- Aspirin (80-100 mg/kg/day) - first-line for arthritis and mild carditis; rapid symptomatic relief (NSAIDs also effective)
- Corticosteroids (prednisolone) - for severe carditis with cardiomegaly or heart failure; no proven benefit over aspirin for long-term valve outcomes
- Haloperidol / valproic acid - for Sydenham chorea
3. Heart failure management if present (diuretics, ACE inhibitors)
4. Activity restriction - limit strenuous exertion in carditis; bed rest not mandatory unless symptomatic
B. Secondary Prophylaxis (Most Important!)
Goal: Prevent recurrent GAS infections and cumulative valve damage
| Indication | Duration |
|---|---|
| ARF without carditis | 5 years or until age 21, whichever is longer |
| ARF with carditis but no residual valve disease | 10 years or until age 21, whichever is longer |
| ARF with persistent valvular disease | 10 years or until age 40 - sometimes lifelong |
Regimen:
- Benzathine penicillin G 1.2 million units IM every 4 weeks (every 3 weeks in high-risk patients) - most effective
- Alternative: Oral penicillin V 250 mg twice daily
- Penicillin-allergic: Oral macrolide (azithromycin 250 mg daily) or oral sulfadiazine
9. Chronic RHD - Valvular Consequences
Mitral Stenosis (Most Common)
- RHD is responsible for 99% of acquired mitral stenosis
- Commissural fusion → narrowed mitral orifice
- Left atrium enlarges → atrial fibrillation → left atrial thrombus → systemic embolism (especially stroke)
- Pulmonary hypertension develops over time
- Classic presentation: dyspnea, hemoptysis, mid-diastolic rumble at apex, opening snap
Mitral Regurgitation
- Can occur acutely (active carditis with valvulitis) or chronically (scarred, retracted leaflets)
Aortic Valve Disease
- Often co-exists with mitral disease
- Regurgitation more common than stenosis from RHD alone
Multi-Valve Disease
- RHD can involve multiple valves simultaneously, compounding hemodynamic compromise
Key Pathology Summary
| Feature | Detail |
|---|---|
| Etiology | Group A Streptococcal pharyngitis → autoimmune cross-reaction |
| Mechanism | Molecular mimicry: anti-M protein antibodies cross-react with cardiac antigens |
| Hallmark lesion | Aschoff body with Anitschkow (caterpillar) cells |
| Acute cardiac lesion | Verrucae (1-2mm) along valve lines of closure; MacCallum plaques |
| Layers affected | Pancarditis (pericarditis, myocarditis, endocarditis) |
| Chronic cardiac lesion | Leaflet thickening, commissural fusion, chordal thickening/fusion |
| Most affected valve | Mitral (65% alone; 25% with aortic) |
| Most important sequela | Mitral stenosis - RHD is the only cause |
| Organisms in lesions | None (sterile - purely immune-mediated) |
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (9780443264528), p. 525-526 | Goldman-Cecil Medicine (9780323930345), p. 855-860 | Braunwald's Heart Disease (9780323722193), p. 856-860 | Fuster and Hurst's The Heart, 15th Ed.
Viva questions on it with answers
Here are high-yield viva questions on Rheumatic Fever and Rheumatic Heart Disease, organized from basic to advanced:
Viva Questions: Rheumatic Fever & Rheumatic Heart Disease
SECTION 1: Basic Concepts
Q1. What is rheumatic fever?
A: Rheumatic fever is an acute, immunologically mediated, multisystem inflammatory disease that occurs 2-3 weeks after Group A Streptococcal (GAS) pharyngitis. It is caused by cross-reactive immune responses (molecular mimicry) - antibodies and T cells directed against streptococcal M proteins also attack cardiac, joint, skin, and neural tissues. It is NOT a direct infection - the heart is sterile.
Q2. What organism causes rheumatic fever? Which site of infection leads to RF?
A: Streptococcus pyogenes - Group A beta-hemolytic Streptococcus (GAS). Only pharyngeal infection (not skin/other sites) classically leads to RF. This is because pharyngeal GAS activates a full humoral + cellular immune response, including T-cell activation, unlike skin infection which primarily activates innate immunity. (Note: emerging evidence suggests skin infections may contribute in some endemic settings like Africa.)
Q3. Why does rheumatic fever occur only in some individuals who get streptococcal pharyngitis?
A: Only 0.3-3% of people with GAS pharyngitis develop RF. This is because:
- Genetic susceptibility is required (heritability ~60%)
- HLA class II associations predispose certain individuals to cross-reactive immune responses
- The proportion of susceptible individuals is fixed at 3-6% globally regardless of geography
- Family history increases risk ~5x
Q4. What is the latent period between streptococcal infection and onset of RF? What is its significance?
A: The latent period is 2-3 weeks. Significance: this is the time required to mount an adaptive immune response (generate cross-reactive antibodies and activated CD4+ T cells). This delay distinguishes RF from direct septic arthritis (which occurs immediately). It also explains why RF can develop even after the throat culture becomes negative.
Q5. Are streptococci found in the heart lesions of RF?
A: No. The heart lesions are completely sterile. This is the key evidence that RF is an immune-mediated (not infectious) disease. The damage is caused by cross-reactive antibodies and T cells, not by direct bacterial invasion.
SECTION 2: Pathogenesis
Q6. What is molecular mimicry? How does it apply to RF?
A: Molecular mimicry is when an immune response generated against a foreign antigen (pathogen) also attacks structurally similar host antigens, causing autoimmune damage. In RF:
- Antibodies against streptococcal M protein cross-react with cardiac myosin
- Antibodies against GAS carbohydrate (GlcNAc) cross-react with laminin in heart valve endothelium
- T cells from peripheral blood and heart valves cross-react with both streptococcal M protein and cardiac myosin
Q7. What is the "two-hit hypothesis" of RF?
A: It proposes that RF valvular disease requires two steps:
- First hit: Cross-reactive antibodies attack valve endothelium, causing inflammation and increased permeability
- Second hit: This facilitates extravasation of cross-reactive T cells through the activated endothelium into valve tissue, leading to formation of Aschoff bodies (granulomatous nodules)
Together, antibody-mediated + T cell-mediated injury causes valvulitis.
Q8. What is the mechanism of Sydenham chorea in RF?
A: Human monoclonal antibodies derived from RF patients target:
- GlcNAc, gangliosides, and dopamine receptors on neuronal cell surfaces in the brain
- These antibodies activate CaMKII (calcium/calmodulin-dependent protein kinase II) in neuronal cells
- They also recognize intracellular tubulin as a biomarker
- This targets the basal ganglia → hyperperfusion and increased metabolism → involuntary movements (chorea)
SECTION 3: Pathology / Morphology
Q9. What is the hallmark histological lesion of rheumatic fever?
A: The Aschoff body (Aschoff-Geipel body) - a focal granulomatous inflammatory nodule found in the myocardium. It consists of:
- A central area of fibrinoid necrosis
- Surrounded by T lymphocytes and occasional plasma cells
- Anitschkow cells (caterpillar cells) - the pathognomonic cell type
Q10. Describe the Anitschkow cell. Why is it called a "caterpillar cell"?
A: Anitschkow cells are plump activated macrophages (histiocytes) with:
- Abundant cytoplasm
- Central, round-to-ovoid nucleus (occasionally binucleate)
- Chromatin that condenses into a central, slender, wavy ribbon - resembling the body of a caterpillar when viewed in longitudinal section
- This ribbon-like chromatin pattern is the reason for the name "caterpillar cell"
- They are pathognomonic of RF and indicate active rheumatic myocarditis
Q11. What is pancarditis? Which layer is most clinically significant?
A: Pancarditis means inflammation of all three layers of the heart:
- Pericarditis - fibrinous; resolves without permanent damage
- Myocarditis - Aschoff bodies in myocardium; rarely causes significant dysfunction
- Endocarditis - most clinically significant; causes valvulitis, verrucae formation, and ultimately leads to chronic RHD
Q12. What are verrucae? Where are they found? How are they different from infective endocarditis vegetations?
A: Verrucae are small (1-2 mm), sterile, warty vegetations that form along the lines of closure of affected valves (mitral > aortic) during acute RF.
| Feature | RF Verrucae | IE Vegetations |
|---|---|---|
| Size | 1-2 mm (small) | Few mm to several cm (large) |
| Location | Lines of closure | Anywhere on valve surface |
| Contents | Sterile fibrin | Bacteria, inflammatory cells |
| Destruction | Minimal initially | Destroys valve tissue |
| Embolic risk | Low | High (septic emboli) |
Q13. What are MacCallum plaques?
A: MacCallum plaques are irregular, rough subendocardial thickenings usually found on the posterior wall of the left atrium. They are caused by regurgitant jets from the incompetent mitral valve repeatedly striking the atrial endocardium during acute RF. They represent a form of jet lesion - endocardial reaction to mechanical injury.
Q14. What are the cardinal pathological changes in the mitral valve in chronic RHD?
A: Four cardinal changes:
- Leaflet thickening (due to fibrosis)
- Commissural fusion - fusion of adjacent leaflet margins → stenosis → "fish-mouth" or "buttonhole" appearance
- Leaflet shortening - leaflets retract
- Thickening and fusion of chordae tendineae - cords become thick, shortened, and fused together
These changes cause mitral stenosis, which is the most important long-term complication of RHD.
Q15. Which valves are affected in RHD and in what order of frequency?
A:
- Mitral alone - ~65-70% (most common)
- Mitral + Aortic - ~25%
- Tricuspid - infrequent (usually combined with mitral)
- Pulmonary - rarely (right-sided valves protected by lower pressures)
The mitral valve is virtually always involved. RHD is the only cause of pure mitral stenosis.
SECTION 4: Clinical Features
Q16. What are the major and minor manifestations of RF? (Jones Criteria)
A:
Major (CASES mnemonic):
- C - Carditis (clinical and/or subclinical echocardiographic valvulitis)
- A - Arthritis (migratory polyarthritis)
- S - Sydenham Chorea (St. Vitus dance)
- E - Erythema marginatum
- S - Subcutaneous nodules
Minor:
- Fever ≥38.5°C
- Elevated ESR/CRP
- Prolonged PR interval on ECG
- Arthralgia (only if arthritis is NOT a major criterion)
Q17. Describe the arthritis of RF. Why is it important that it does NOT cause permanent damage?
A: Characteristics:
- Migratory polyarthritis (moves from joint to joint, not additive)
- Affects large joints - knees, ankles, wrists, elbows (rarely hips and spine)
- Extremely painful, warm, swollen
- Synovial fluid is sterile with lymphocyte predominance
- Responds rapidly to aspirin/NSAIDs - failure to respond should prompt reconsideration of diagnosis
- Resolves completely in ~1 month without any residual joint damage
Clinically important: although the most painful and obvious manifestation, the arthritis is the least dangerous. The carditis is silent but causes permanent damage.
Q18. What is Sydenham chorea? What are its clinical features?
A: Sydenham chorea (St. Vitus dance) occurs in ~30% of RF patients. Features:
- Involuntary, non-rhythmic, purposeless movements of body, limbs, and face
- Usually more pronounced on one side (hemichorea)
- Disappears during sleep - distinguishes it from tics/tremors
- Milkmaid grip - inability to maintain sustained grip
- Pronator sign - when arms raised, hands pronate
- May occur in isolation weeks to months after the acute strep infection (latent period up to 6 months)
- Usually resolves in months; ~1/3 have recurrences
- Treated with haloperidol, valproic acid, or carbamazepine
Q19. What is erythema marginatum?
A: A characteristic but uncommon (<10%) skin rash of RF:
- Flat or slightly raised, non-pruritic, non-scarring pink/red macules or papules
- Spreads in a serpiginous (snake-like) pattern, expanding outward with central clearing
- Located on the trunk and proximal limbs (NOT the face)
- Evanescent - can move and change hour to hour (the pen mark in clinical photos shows it moves ~60 min later)
- Does NOT scar
- Histology: perivascular neutrophilic and mononuclear infiltrates of the dermis
Q20. What is the frequency of carditis in RF? Which valve lesion occurs first - stenosis or regurgitation?
A: Carditis occurs in >50% of RF patients. In acute RF, valvulitis causes regurgitation first (not stenosis). Stenosis is a chronic change that develops over years after repeated attacks due to progressive commissural fusion. A child with acute RF will have a mitral regurgitation murmur; the stenotic "fish-mouth" valve develops only with chronic RHD.
SECTION 5: Diagnosis
Q21. What is the 2015 revision of the Jones Criteria? What are the key changes from older versions?
A: Three key changes in 2015 AHA revision:
- Subclinical valvulitis on echocardiography accepted as a major criterion (even without clinical murmur)
- Risk stratification into low-risk vs. moderate/high-risk populations with different criteria thresholds
- In moderate/high-risk populations: monoarthritis and polyarthralgia upgraded to major criteria; monoarthralgia added as minor criterion
For diagnosis of initial ARF: 2 major OR 1 major + 2 minor criteria (PLUS evidence of preceding GAS infection)
Q22. What evidence of preceding GAS infection is required for Jones Criteria?
A:
- Positive throat swab culture for GAS
- Positive rapid streptococcal antigen test
- Elevated or rising ASO (Anti-Streptolysin O) titer - most commonly used
- Elevated anti-DNase B titer
- Note: in up to 1/3 of patients, no symptoms of preceding pharyngitis are reported - subclinical infection
Q23. Why is echocardiography important in ARF?
A: Echocardiography is recommended for ALL patients with confirmed or suspected ARF because:
- Detects subclinical carditis in patients with no murmurs (now a major criterion)
- Assesses severity of valvular involvement
- Monitors patients with isolated Sydenham chorea (carditis may be silent)
- Establishes baseline for monitoring RHD progression
- Monitors recurrences
SECTION 6: Treatment and Prophylaxis
Q24. What are the 4 primary goals of treatment in ARF?
A:
- Suppress inflammation - minimize cardiac and joint damage
- Eradicate GAS from the pharynx (even if culture negative)
- Relieve symptoms - especially pain and fever
- Initiate secondary prophylaxis - prevent future attacks and cumulative valve damage
Q25. What antibiotic is used to eradicate GAS in ARF? What is the dose?
A: Drug of choice: Benzathine Penicillin G - single IM injection (1.2 million units). Alternatives:
- 10-day course of oral penicillin or amoxicillin
- Erythromycin if penicillin-allergic
This eradicates any residual GAS even though throat swabs are often already negative by the time ARF is diagnosed.
Q26. What is secondary prophylaxis in RF? What is its duration?
A: Secondary prophylaxis means long-term antibiotic treatment to prevent recurrent GAS pharyngitis (which would trigger further RF attacks and cumulative valve damage).
Regimen: Benzathine penicillin G 1.2 million units IM every 4 weeks (every 3 weeks in high-risk)
Duration:
| Situation | Duration |
|---|---|
| RF without carditis | 5 years or until age 21 (whichever longer) |
| RF + carditis, no residual valve disease | 10 years or until age 21 |
| RF + persistent valvular disease | 10 years or until age 40 - sometimes lifelong |
Q27. Why is continuous secondary prophylaxis more important than treating the acute attack?
A: Because:
- Each recurrence of ARF causes cumulative, additive valve damage
- ~50% of patients who have another untreated GAS infection get a new RF attack
- The chronic valvular disease (especially mitral stenosis) that causes death develops over years from repeated attacks
- Preventing recurrences is the single most effective intervention to reduce RHD mortality
- Even patients who recovered fully from acute RF with no valve disease can develop chronic RHD from subsequent attacks
SECTION 7: High-Yield / Tricky Questions
Q28. RHD is the only cause of which valve lesion?
A: Acquired mitral stenosis - RHD accounts for virtually 100% (99%) of cases. Other valve lesions (aortic stenosis, aortic regurgitation, mitral regurgitation) have multiple causes. But pure or dominant mitral stenosis = RHD until proven otherwise.
Q29. A student says "RF causes mitral stenosis acutely." Is this correct?
A: No - this is a common error. During acute RF, valvulitis causes mitral regurgitation (the inflamed, edematous leaflets fail to coapt properly). Mitral stenosis is a chronic change developing over years to decades as a result of progressive fibrosis, commissural fusion, and chordal scarring from repeated acute attacks. A child presenting with first-episode RF will have a regurgitant murmur, not stenosis.
Q30. If a patient has RF with severe carditis, should you give corticosteroids? Will they prevent valve damage?
A: Corticosteroids (prednisolone) are indicated for severe carditis with cardiomegaly or heart failure to suppress inflammation rapidly. However, there is no proven benefit over aspirin for preventing long-term valvular damage or altering the progression to chronic RHD. The key determinant of long-term outcome is prevention of recurrences through secondary prophylaxis, not the anti-inflammatory drug used in the acute attack.
Q31. What is the difference between RF arthritis and septic arthritis on synovial fluid analysis?
A: In RF arthritis, synovial fluid is sterile with lymphocyte predominance - no organisms on culture or Gram stain. In septic arthritis, organisms are present with neutrophil predominance. Also, RF arthritis responds dramatically to aspirin/NSAIDs (septic arthritis does not improve with anti-inflammatories alone and requires antibiotics/drainage).
Q32. Name the diseases that can mimic RF and must be excluded (differential diagnosis).
A:
- Juvenile Idiopathic Arthritis (JIA) - especially systemic type (Still disease); distinguished by diurnal fever, evanescent rash, prolonged joint symptoms
- Lyme disease - Borrelia arthritis
- Reactive arthritis (post-infectious)
- Infective endocarditis - fever + valve lesions
- SLE - Libman-Sacks endocarditis
- Kawasaki disease (in children)
- Sickle cell arthropathy
- Leukemia/lymphoma - can present with joint pain and fever
- Serum sickness
- Gonococcal arthritis (in adolescents)
Q33. What is the prognosis of RF/RHD?
A:
- Murmurs resolve in ~50% of patients with mild carditis and no major cardiac enlargement
- Without secondary prophylaxis, chronic RHD develops in a significant proportion
- Chronic RHD is the cause of 25-45% of all cardiovascular disease in developing countries and a major cause of heart failure
- Complications of established RHD include: atrial fibrillation, left atrial thrombus, systemic embolism (stroke), pulmonary hypertension, heart failure, and infective endocarditis
- Valve repair or replacement may eventually be required
High-Yield Examiner Favorites:
- Anitschkow cell = caterpillar cell = pathognomonic of RF
- Aschoff body = pathognomonic myocardial granuloma
- RHD = only cause of mitral stenosis
- Acute RF → mitral REGURGITATION; Chronic RHD → mitral STENOSIS
- Streptococci absent from heart lesions (immune-mediated, not infectious)
- Benzathine penicillin IM monthly = secondary prophylaxis cornerstone
Pharmacology; hematinics
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
I now have comprehensive pharmacology content from Goodman & Gilman's. Let me compile the full answer.
Pharmacology of Hematinics
Hematinics are drugs and substances that provide raw materials necessary for erythropoiesis - the production of red blood cells. They include iron, vitamin B12 (cyanocobalamin), folic acid, and erythropoiesis-stimulating agents (ESAs).
PART 1: IRON
Iron in the Body
Total body iron = 3-5 g (male ~50 mg/kg, female ~37 mg/kg)
| Compartment | Amount | Form |
|---|---|---|
| Hemoglobin | ~2.5 g (largest) | Heme iron in RBCs |
| Myoglobin + enzymes | ~300-400 mg | Heme/non-heme |
| Storage (ferritin/hemosiderin) | ~1 g (male), ~0.3 g (female) | Liver, spleen, bone marrow |
| Transport (transferrin) | ~4 mg | Plasma |
Iron is essential for:
- Oxygen transport (hemoglobin, myoglobin)
- Electron transport chain (cytochromes)
- DNA synthesis
- Enzyme function (catalase, peroxidase, xanthine oxidase)
Iron Metabolism and Absorption
Normal daily turnover: ~1 mg/day (lost via desquamated gut/skin cells, sweat)
Daily requirements:
- Adult male: ~1 mg/day
- Menstruating female: ~1.5-2 mg/day
- Pregnant female: ~3-4 mg/day
- Children/adolescents: +0.5 mg/day above adult
Dietary iron intake: ~20-30 mg/day in meat-eaters; only ~10% (1-2 mg) absorbed normally
Mechanism of Iron Absorption (Step by Step)
Site: Primarily proximal duodenum and upper jejunum
Dietary iron (mainly Fe³⁺)
↓ Gastric acid + Duodenal cytochrome b (DcytB) — ferrireductase
Reduced to Fe²⁺
↓ DMT1 (Divalent Metal Transporter-1) on apical membrane
Enters enterocyte
↓ Stored as ferritin (if excess) OR exported via
Ferroportin-1 (FPN1) on basolateral membrane
↓ Hephaestin (ferroxidase) — re-oxidizes Fe²⁺ → Fe³⁺
Binds apotransferrin in plasma → Transferrin-bound iron
↓
Delivered to bone marrow, liver, other tissues
Heme iron (from meat - hemoglobin/myoglobin):
- Absorbed as intact heme moiety via Heme Carrier Protein-1
- More bioavailable than non-heme iron
- Once inside enterocyte, heme oxygenase releases Fe²⁺
Factors ENHANCING absorption:
- Ascorbic acid (Vitamin C) - reduces Fe³⁺ → Fe²⁺
- Acidic pH (gastric acid)
- Animal protein (heme iron)
- Iron deficiency state (upregulates DMT1, FPN1)
- Hypoxia
Factors REDUCING absorption:
- Phytates (cereals, legumes)
- Tannins (tea, coffee)
- Oxalates, phosphates
- Antacids, PPIs (raise gastric pH)
- Tetracyclines, fluoroquinolones (chelate iron)
- Calcium (competes with DMT1)
- Excess iron (hepcidin mediated)
Hepcidin - Master Regulator of Iron
Hepcidin is a peptide hormone produced by the liver. It is the central regulator of iron homeostasis:
- Binds to Ferroportin-1 (FPN1) → causes its internalization and degradation
- This blocks iron export from enterocytes, macrophages, and hepatocytes
- Net effect: reduces circulating iron
Hepcidin rises with: iron overload, inflammation (IL-6) → explains anemia of chronic disease (hepcidin traps iron in macrophages, makes it unavailable for erythropoiesis)
Hepcidin falls with: iron deficiency, hypoxia, increased erythropoietic demand → increases iron absorption
Iron Deficiency Anemia (IDA)
Most common nutritional anemia globally. Causes: inadequate intake, malabsorption, blood loss (GI, menstrual), increased demand (pregnancy).
Lab findings:
- Low serum iron
- Low ferritin (first to fall - most sensitive early marker)
- High TIBC (Total Iron Binding Capacity)
- Low transferrin saturation (<16%)
- Microcytic, hypochromic RBCs on smear
- Low MCV (<80 fL)
Non-hematological symptoms of IDA:
- Fatigue, koilonychia (spoon nails), angular stomatitis, glossitis
- Pica (craving for ice, clay, starch)
- Restless legs syndrome
- Impaired cognitive/behavioral development in children
Oral Iron Preparations
| Preparation | Elemental Iron Content | Notes |
|---|---|---|
| Ferrous sulfate (FeSO₄) | 20% (65 mg per 325 mg tablet) | Cheapest, most widely used |
| Ferrous gluconate | 12% | Better GI tolerance |
| Ferrous fumarate | 33% | Highest elemental iron % |
| Ferrous ascorbate | Variable | Ascorbate enhances absorption |
| Carbonyl iron | Near 100% | Slow-release; lower toxicity in overdose |
Why ferrous (Fe²⁺) over ferric (Fe³⁺)?
- Fe²⁺ is more soluble at physiologic pH
- Directly transported by DMT1
- Fe³⁺ must first be reduced to Fe²⁺ before absorption
Standard adult dose: Ferrous sulfate 325 mg three times daily (= 195 mg elemental iron/day)
Pediatric dose: 2 mg/kg/day elemental iron orally
Administration tips:
- Take on empty stomach (better absorption)
- Take with Vitamin C (enhances absorption)
- Avoid with milk, antacids, tea, coffee, tetracyclines
- If GI intolerance, take with food (reduces absorption ~40%)
Monitoring response:
- Reticulocytosis appears in 3-4 days (children) or >1 week (adults) - first sign of response
- Hemoglobin rises by 1-2 g/dL per month
- Continue 3-6 months AFTER hemoglobin normalizes to replenish stores
Adverse effects:
- Nausea, vomiting, epigastric discomfort
- Constipation (most common) or diarrhea
- Black/dark stools (warn patients - can mimic melena)
- Staining of teeth with liquid preparations (use straw)
Parenteral Iron Preparations
Indicated when:
- Oral iron not tolerated
- Malabsorption (celiac, IBD, post-gastrectomy)
- Chronic kidney disease (CKD) - hepcidin blocks oral absorption
- Non-compliance
- Rapid iron repletion needed (e.g. pre-surgery)
- Chronic blood loss exceeding oral replacement capacity
| Preparation | Route | Dose | Notes |
|---|---|---|---|
| Iron dextran (LMW) | IV/IM | Up to 1000 mg single dose | Highest anaphylaxis risk; test dose required; avoid IM (necrosis risk) |
| Ferric gluconate | IV | 125-250 mg per session | Safer than dextran; 80% delivered to transferrin within 24h |
| Iron sucrose | IV | 100-300 mg per session | Well tolerated; approved in CKD, pregnancy |
| Ferric carboxymaltose | IV | 750 mg x2 doses | High-dose single infusion possible |
| Ferumoxytol | IV | 1020 mg single dose | Fastest; risk of hypotension; black box warning |
| Iron isomaltoside | IV | Up to 1000 mg single dose | Stable complex; low anaphylaxis |
Iron dextran - most important adverse effects:
- Anaphylaxis (serious - requires test dose + monitoring for 1 hour post-infusion)
- Delayed hypersensitivity: fever, malaise, arthralgia, lymphadenopathy (days-weeks later)
- IM injection risk: local pain, tissue necrosis, potential malignant change at injection site - IM route now largely abandoned
- Use extreme caution in rheumatoid arthritis (increased risk of delayed reactions)
- Withhold if plasma ferritin >800 μg/L (iron overload risk)
Iron Toxicity / Overdose
Acute iron poisoning (commonly in children ingesting adult tablets):
- Phases: GI symptoms → apparent recovery → shock and hepatic failure → recovery or death
- Mechanism: Fe²⁺ generates free radicals (Fenton reaction) → lipid peroxidation → cell death
- Treatment: Gastric lavage + Deferoxamine (chelation therapy) - binds free iron → excreted as ferrioxamine in urine (pink-orange discoloration)
PART 2: FOLIC ACID (Vitamin B9)
Biochemistry
Dietary form: Polyglutamate folates (in food)
Pharmaceutical form: Pteroylglutamic acid (PteGlu) - monoglutamate
After absorption → reduced to Tetrahydrofolate (THF) → acts as one-carbon carrier for:
| Reaction | Coenzyme needed | Significance |
|---|---|---|
| Homocysteine → Methionine | 5-CH₃-THF + Vit B12 | Links folate and B12 |
| dUMP → dTMP (thymidylate) | 5,10-CH₂-THF | DNA synthesis - rate-limiting step |
| Purine synthesis (C-2, C-8) | 10-CHO-THF | DNA/RNA synthesis |
| Serine → Glycine | THF | One-carbon metabolism |
| Histidine metabolism | H₄PteGlu | FIGLU → glutamic acid |
Key point: Folate deficiency primarily impairs DNA synthesis → affects rapidly dividing cells (bone marrow, GI epithelium, fetus) → megaloblastic anemia + neural tube defects
Dietary Sources and Requirements
- Rich sources: fresh green vegetables (spinach, broccoli, asparagus), liver, yeast, fruits
- Cooking destroys up to 90% of folate
- Daily requirement: 400 μg/day (adults); 600 μg/day (pregnancy); 500 μg/day (lactation)
- Body stores: ~5-20 mg (liver) - depleted in 3-4 months of deficiency (compared to years for B12)
Causes of Folate Deficiency
- Inadequate intake - poor diet, alcoholism (most common cause)
- Increased demand - pregnancy, hemolytic anemia, rapid cell turnover
- Malabsorption - celiac disease, tropical sprue
- Drug-induced - methotrexate (DHFR inhibitor), trimethoprim, phenytoin, sulfasalazine
- Alcohol - impairs absorption AND utilization
Pharmacological Uses of Folic Acid
1. Megaloblastic anemia due to folate deficiency:
- Dose: 5 mg/day orally for 4 months (or until dietary issues corrected)
- Parenteral if malabsorption
2. Prevention of Neural Tube Defects (NTD):
- Folic acid supplements reduce NTD risk by ~50-70%
- Periconceptional supplementation: 400 μg/day starting 1 month BEFORE conception and for first 12 weeks
- High-risk (previous NTD child): 4-5 mg/day
3. Pregnancy supplementation:
- 600 μg/day throughout pregnancy
4. Megaloblastic anemia from methotrexate:
- Use folinic acid (leucovorin/citrovorum factor) - bypasses DHFR inhibition
- This is the "leucovorin rescue" in cancer chemotherapy
Important Warning: Folate vs. B12 in Megaloblastic Anemia
NEVER give folic acid alone in suspected B12 deficiency without ruling out B12 deficiency first!
- Folic acid corrects the hematological picture (anemia) in B12 deficiency
- BUT it does NOT prevent or may worsen the neurological damage (subacute combined degeneration of the spinal cord)
- Always check both folate AND B12 levels before treating megaloblastic anemia
PART 3: VITAMIN B12 (Cobalamin)
Forms
- Cyanocobalamin - stable, synthetic, pharmaceutical form; converted to active forms in body
- Hydroxocobalamin - injectable; longer lasting (preferred for IM); better tissue retention
- Methylcobalamin - active form in cytoplasm
- Adenosylcobalamin - active form in mitochondria
Biochemical Roles
1. Methionine synthesis (cytoplasm):
Homocysteine + 5-CH₃-THF → Methionine + THF
- Cofactor: Methylcobalamin
- Failure → "methyl-folate trap" → functional folate deficiency → megaloblastic anemia
- Elevated homocysteine → cardiovascular risk
2. Methylmalonyl-CoA → Succinyl-CoA (mitochondria):
- Cofactor: Adenosylcobalamin
- Failure → methylmalonic acid (MMA) accumulates → myelin damage → subacute combined degeneration of spinal cord
- MMA is a specific marker of B12 deficiency (elevated in B12 but NOT folate deficiency)
Absorption of B12 (Critical Pathway)
Dietary B12 (bound to food proteins)
↓ Pepsin + HCl (stomach)
Released from food
↓ Binds R-protein (haptocorrin, from saliva/gastric juice)
B12-R complex
↓ Pancreatic proteases in duodenum degrade R-protein
Free B12
↓ Binds Intrinsic Factor (IF) secreted by gastric parietal cells
B12-IF complex
↓ Absorbed at specific receptors in TERMINAL ILEUM (cubilin receptor)
Enters portal blood
↓ Binds transcobalamin II (TC II) for transport to tissues
Intrinsic Factor (IF) is the critical limiting factor - its absence = pernicious anemia
Daily requirement: 2 μg/day
Body stores: 2-5 mg (mainly liver) - lasts 3-5 years after absorption stops
Causes of B12 Deficiency
| Cause | Mechanism |
|---|---|
| Pernicious anemia | Autoimmune - anti-parietal cell / anti-IF antibodies → no IF |
| Strict veganism | No dietary animal products |
| Gastrectomy | Loss of parietal cells → no IF |
| Terminal ileum disease/resection | (Crohn's, resection) - no absorption site |
| Pancreatic insufficiency | Can't digest R-protein in duodenum |
| Fish tapeworm (D. latum) | Competes for B12 in gut |
| Metformin (long-term) | Reduces B12 absorption (Ca-dependent mechanism) |
| Nitrous oxide | Irreversibly oxidizes methylcobalamin |
Clinical Features of B12 Deficiency
- Megaloblastic anemia - hypersegmented neutrophils (>5 lobes), macro-ovalocytes, pancytopenia
- Neurological: Subacute Combined Degeneration (SCD) of spinal cord:
- Posterior columns (vibration, proprioception loss)
- Lateral columns (UMN signs - spasticity, hyperreflexia)
- Peripheral neuropathy
- Dementia, psychiatric symptoms
- Glossitis (beefy red, smooth tongue)
- Elevated homocysteine AND methylmalonic acid (diagnostic)
Treatment of B12 Deficiency
Pernicious anemia / malabsorption (cannot absorb orally):
- Hydroxocobalamin 1000 μg IM - loading: daily for 1 week, then weekly for 4 weeks, then monthly for life
- Cyanocobalamin 1000 μg IM as alternative
Dietary deficiency (absorption intact):
- Oral cyanocobalamin 1000-2000 μg/day - effective even without IF through passive diffusion (~1% absorbed passively)
- Intranasal cyanocobalamin available for maintenance
Response monitoring:
- Reticulocytosis peaks at 5-7 days
- Hemoglobin normalizes in 6-8 weeks
- Neurological improvement begins in weeks but may be incomplete if severe
PART 4: ERYTHROPOIESIS-STIMULATING AGENTS (ESAs)
Used when anemia is due to inadequate erythropoietin (EPO) production (not iron/vitamin deficiency).
| Drug | Type | Route | Indication |
|---|---|---|---|
| Epoetin alfa (rHuEPO) | Recombinant EPO | SC/IV | CKD, chemotherapy-induced anemia |
| Darbepoetin alfa | Hyperglycosylated EPO - longer t½ | SC/IV | CKD, chemotherapy-induced anemia |
| Methoxy-PEG-epoetin beta (CERA) | Continuous EPO receptor activator | SC/IV | CKD |
Mechanism: Bind EPO receptor on erythroid progenitors → activate JAK2-STAT5 → promote survival, proliferation, differentiation of RBC precursors
Target Hb: 10-12 g/dL in CKD (avoid targeting >13 g/dL - increased stroke/thrombosis risk)
Adverse effects:
- Hypertension (increased blood viscosity)
- Thrombosis/stroke (especially if over-correction)
- Pure red cell aplasia (anti-EPO antibodies - rare)
- Iron deficiency develops rapidly (increased demand) - must supplement iron concurrently
SUMMARY TABLE: Hematinics at a Glance
| Drug | Deficiency State | Type of Anemia | Key Mechanism | Key Monitoring |
|---|---|---|---|---|
| Ferrous sulfate (oral) | Iron deficiency | Microcytic hypochromic | Replaces iron stores | Reticulocytosis at 1 wk; Hb at 4 wks; continue 3-6 months |
| Parenteral iron | IDA with oral intolerance/malabsorption | Microcytic hypochromic | Rapid iron repletion | Ferritin; risk of anaphylaxis with dextran |
| Folic acid | Folate deficiency | Megaloblastic | One-carbon donor for DNA synthesis | Rule out B12 deficiency FIRST |
| Cyanocobalamin/Hydroxocobalamin | B12 deficiency | Megaloblastic + neurological | Methionine synthesis + myelin maintenance | MMA, homocysteine levels; neurological recovery |
| EPO/Darbepoetin | CKD, chemotherapy | Normocytic (EPO deficiency) | Stimulate erythroid progenitors | Hb target 10-12 g/dL; BP; iron stores |
Sources: Goodman & Gilman's Pharmacological Basis of Therapeutics 13th Ed., Ch. 45 | Sleisenger & Fordtran's GI and Liver Disease | Rosen's Emergency Medicine | Comprehensive Clinical Nephrology 7th Ed.
This is a shared conversation. Sign in to Orris to start your own chat.