In acute ischemic stroke in brain when to control and on what day should we consider decreasing bp
Reading File
Finding Sources
Finding Sources
Searching PubMed
"ischemic stroke" AND "blood pressure" AND management
Reading File
Reading File
Finding Sources
Reading Article
PMID: 38386320
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive data from multiple textbooks plus the clinical flowchart. Let me compile the full answer.
BP Management in Acute Ischemic Stroke (AIS)
The key principle is permissive hypertension in most patients - BP elevation after stroke is a protective physiological response that maintains perfusion to the ischemic penumbra. Aggressive BP lowering in the acute phase can extend the infarct. Management depends entirely on whether the patient is a thrombolysis/thrombectomy candidate.
When NOT to Treat BP (Permissive Hypertension)
For patients not eligible for IV thrombolysis or thrombectomy, current AHA guidelines recommend treating only if:
- Systolic BP > 220 mmHg, OR
- Diastolic BP > 120 mmHg
Even then, the goal is a 10-15% reduction only - not normalizing BP. Both hypo- and hypertension in the acute phase are associated with poor outcome (U-shaped relationship). Low BP (SBP <155, DBP <70, or MAP <100 mmHg) in the first few hours correlates with increased 90-day mortality.
When to ACTIVELY Control BP
1. Patient is a thrombolysis (rtPA) candidate
- BP must be brought to < 185/110 mmHg BEFORE giving IV rtPA
- If BP cannot be maintained at or below 185/110, do NOT administer rtPA
- After rtPA infusion: maintain BP < 180/105 mmHg for the first 24 hours
- Monitor every 15 min for 2 hours from start, then every 30 min for 6 hours, then every hour for 16 hours
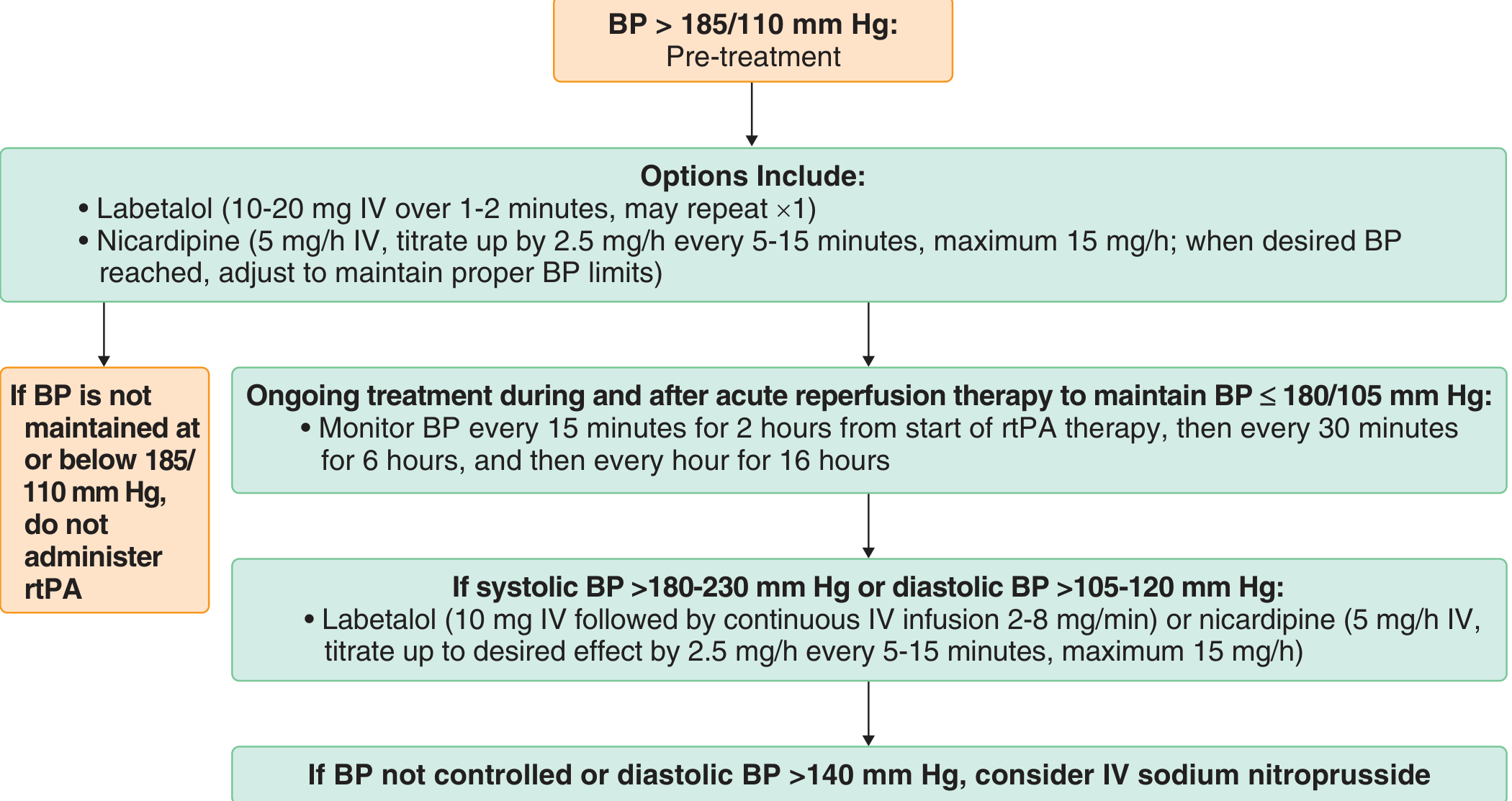
2. After endovascular thrombectomy (EVT)
- After successful recanalization/reperfusion, maintain BP < 180/105 mmHg in the first 24 hours
- After complete reperfusion (TICI 3), some centers lower to SBP 120-140 mmHg to minimize reperfusion injury/hyperperfusion syndrome
- Important 2024 evidence: A JAMA Network Open meta-analysis (Ghozy et al., 2024) (4 RCTs, n=1571) found that intensive BP reduction post-EVT leads to LOWER functional independence (RR 0.81, 95% CI 0.67-0.98) without reducing mortality or hemorrhage - so conservative BP management is currently favored after thrombectomy.
3. Diastolic BP > 140 mmHg (any patient)
- Use IV sodium nitroprusside infusion
Drug Choices for Acute BP Reduction
| Situation | Agent | Dose |
|---|---|---|
| SBP 180-230 or DBP 105-120 | Labetalol IV | 10-20 mg over 1-2 min, may repeat x1; or infusion 2-8 mg/min |
| SBP 180-230 or DBP 105-120 | Nicardipine IV | 5 mg/h, titrate up by 2.5 mg/h every 5-15 min, max 15 mg/h |
| DBP > 140 | Sodium nitroprusside IV | Continuous infusion |
| General | Hydralazine IV | Intermittent bolus |
When to Start Decreasing BP for Secondary Prevention (Subacute Phase)
This is a commonly asked clinical question. The answer is after 24-48 hours for most patients, or more commonly after 3-7 days, depending on stability:
- The spontaneous BP elevation after stroke tends to decline naturally over the first 3-7 days in most patients
- Most guidelines and trial evidence suggest restarting or initiating antihypertensive therapy around day 2-3 onwards in clinically stable patients, targeting SBP < 140 mmHg for long-term secondary prevention
- The COSSACS trial showed that continuing pre-existing antihypertensives acutely did NOT reduce 2-week death or dependency - so there is no urgency to restart them in the immediate 24-hour window
- For long-term prevention, antihypertensives (particularly ACE inhibitors + thiazides) are resumed during the hospitalization once the patient is neurologically stable, typically by day 3-7
- In patients with hemorrhagic conversion, BP control becomes more aggressive from that point
Summary Table
| Patient Category | BP Threshold to Treat | Target | Timing to Reduce |
|---|---|---|---|
| No thrombolysis/EVT | > 220/120 mmHg | Reduce by 10-15% | During acute phase only if extreme |
| Pre-thrombolysis | > 185/110 mmHg | < 185/110 before rtPA | Immediate (to qualify for rtPA) |
| Post-thrombolysis | > 180/105 mmHg | < 180/105 | First 24 hours |
| Post-EVT (incomplete recanalization) | > 180/105 mmHg | < 180/105 | First 24 hours |
| Post-EVT (complete reperfusion) | Permissive | 120-140 mmHg SBP | Immediately after |
| Secondary prevention (all) | Any elevation | < 140/90 mmHg | Day 2-7, once stable |
Key takeaway: Permissive hypertension is the rule in the first 24 hours unless the patient is receiving thrombolysis/thrombectomy or has extreme hypertension (>220/120). Start downward titration toward secondary prevention targets from approximately day 2-3 onward when the patient is neurologically stable, completing the transition by day 7 in most cases.
- Bradley and Daroff's Neurology in Clinical Practice, p. 1122
- Plum and Posner's Diagnosis and Treatment of Stupor and Coma, p. 595-596
- Comprehensive Clinical Nephrology 7e, p. 581
- Ghozy et al., JAMA Network Open 2024 - Intensive vs Conventional BP Control After Thrombectomy
Neuromyeletis optica spectrum disorder
Reading File
Finding Sources
Finding Sources
Searching PubMed
"neuromyelitis optica spectrum disorder" AND treatment
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have very comprehensive data from multiple authoritative sources. Let me compile the full structured answer.
Neuromyelitis Optica Spectrum Disorder (NMOSD)
Overview / History
NMOSD (formerly called Devic disease, named after the French physician Eugene Devic, 1894) is an aggressive, antibody-mediated autoimmune inflammatory disorder of the CNS with a predilection for the optic nerves and spinal cord. It was considered a variant of multiple sclerosis (MS) until 2004, when the aquaporin-4 (AQP4) antibody was discovered, firmly establishing NMOSD as a distinct disease entity.
Pathophysiology
The central target is the aquaporin-4 (AQP4) water channel protein, which is heavily expressed on astrocytic end-feet at the blood-brain barrier, periependymal areas, and around the central canal of the spinal cord.
Mechanism of injury:
- AQP4-IgG (NMO-IgG) binds to AQP4 on astrocyte foot processes
- This activates the complement cascade (C5-C9 membrane attack complex) via antibody-dependent cell-mediated cytotoxicity
- Results in a distinctive "rim and rosette" pattern of immune complex deposition
- Leads to astrocyte death and tissue necrosis affecting both gray and white matter
- Inflammatory infiltrate includes T cells, B cells, macrophages, neutrophils, and eosinophils (distinguishing it from MS where neutrophils are uncommon)
- Secondary demyelination follows the primary astrocytopathy
- Vascular immunoglobulin and complement deposition is seen in damaged white matter
- Loss of AQP4 expression on immunohistochemistry is a hallmark pathological finding
In contrast to MS: NMOSD is a humoral (antibody-mediated) disease vs. the primarily T-cell driven pathology of MS.
Epidemiology
| Feature | NMOSD | MS (for comparison) |
|---|---|---|
| Prevalence | 0.5-4.4/100,000 | 50-100x more common |
| Female:Male ratio | 9:1 (AQP4+) | ~3:1 |
| Age of onset | ~40 years (median) | ~28 years |
| Racial predisposition | Non-Caucasian predominance (Asian, African, Hispanic) | Caucasian predominance |
| Course | Relapsing (85%), rarely progressive | Often secondary progressive |
| Disability | Severe from relapses (blindness, paralysis) | Accumulates more gradually |
Core Clinical Characteristics (6 Domains)
According to the 2015 International Panel criteria, there are 6 core clinical characteristics:
- Optic neuritis (ON) - often severe, bilateral, with poor recovery; affects >1/2 optic nerve length or involves optic chiasm
- Acute myelitis - longitudinally extensive transverse myelitis (LETM) spanning ≥3 contiguous vertebral segments
- Area postrema syndrome - intractable nausea, vomiting, hiccups (presenting symptom in ~10%); due to lesions in dorsal medulla
- Acute brainstem syndrome - double vision, dysphagia, oculomotor dysfunction, ataxia, respiratory compromise
- Symptomatic narcolepsy or acute diencephalic syndrome - associated with hypothalamic lesions; may cause inappropriate diuresis, hypersomnia, anorexia, hypothermia
- Symptomatic cerebral syndrome with NMOSD-typical brain lesions
MRI Features
Spinal cord (most characteristic):
- LETM: T2 hyperintense lesions spanning ≥3 contiguous vertebral segments, often extending from cervical junction throughout cervical and thoracic cord
- Lesions centered on the gray matter (axial view)
- Areas of focal swelling and enhancement in acute phase
- Focal cord atrophy in chronic phase
Brain MRI (often affected despite earlier belief otherwise):
- Lesions parallel sites of high AQP4 expression (circumventricular organs)
- Periependymal regions surrounding 3rd and lateral ventricles
- Hypothalamus, corpus callosum, dorsal brainstem/pons (adjacent to 4th ventricle)
- Area postrema / dorsal medulla
- Large hemispheric lesions with a "cloud-like" appearance that can resolve completely (unlike MS)
- Corticospinal tract lesions
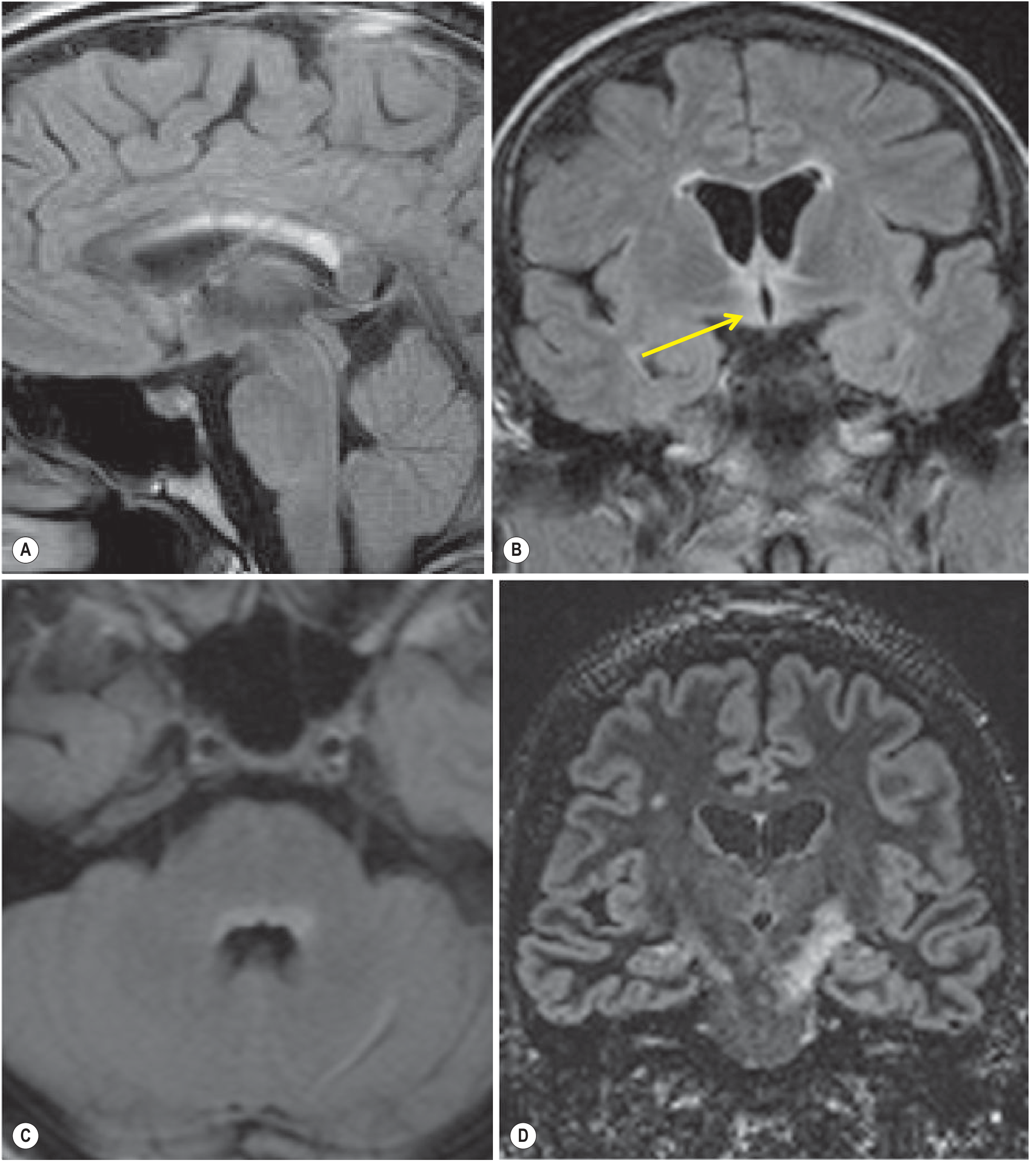
Optic nerve MRI:
- T2 hyperintense or gadolinium-enhancing lesion extending >1/2 optic nerve length or involving the optic chiasm
Diagnostic Criteria (2015 International Panel - IPND)
AQP4-IgG Seropositive NMOSD
- At least one core clinical characteristic
- Positive AQP4-IgG (cell-based assay strongly recommended)
- Exclusion of alternative diagnoses
AQP4-IgG Seronegative (or Unknown Status)
- At least two core clinical characteristics from one or more attacks, meeting ALL of:
- a. At least one must be optic neuritis, LETM myelitis, or area postrema syndrome
- b. Dissemination in space (≥2 different core characteristics)
- c. Fulfillment of additional MRI requirements (as listed above)
- Negative AQP4-IgG (or unavailable)
- Exclusion of alternative diagnoses
CSF Findings
| Feature | NMOSD | MS |
|---|---|---|
| Pleocytosis | Often >50 leukocytes/mm³ | Usually <50 |
| Cell type | Neutrophils + eosinophils present | Predominantly lymphocytes |
| Oligoclonal bands (OCBs) | <20% (uncommon) | 80-90% positive |
| AQP4-IgG in CSF | May be positive | Negative |
Serology: AQP4-IgG vs MOG-IgG
| Feature | AQP4-IgG Positive (~70-80% of NMOSD) | MOG-IgG Positive |
|---|---|---|
| Female predominance | Strong (9:1) | Equal sex distribution |
| Age of onset | Late 4th decade | Younger (children/young adults) |
| Course | Mostly relapsing, severe | Often monophasic or serial monophasic |
| Outcome | Poor recovery, high disability | More favorable |
| ASSociated autoimmune diseases | Yes (Sjögren, SLE, etc.) | Less common |
| Link with ADEM | No | Yes (ADEM is most common MOGAD presentation) |
| Response to approved biologics | Yes | No FDA-approved agents yet |
Key Differentiators from MS
| Feature | NMOSD | MS |
|---|---|---|
| Primary target | Astrocyte (AQP4) | Oligodendrocyte/myelin |
| Optic neuritis | Severe, often bilateral, poor recovery | Usually unilateral, good recovery |
| Myelitis | Longitudinally extensive (≥3 segments) | Short segment (<2 segments) |
| Progressive course | Rare | Common (secondary progressive) |
| OCBs in CSF | <20% | 80-90% |
| Response to MS DMTs | MS drugs (interferons, natalizumab) may WORSEN NMOSD | Effective |
| Pathology | Astrocytopathy + necrosis | Demyelinating plaques |
Treatment
Acute Attack Management
- High-dose IV methylprednisolone: 1 g/day for 5-10 days, followed by oral prednisone taper
- Plasma exchange (PLEX): 5-7 exchanges of 1.5 plasma volumes each - used for attacks not responding to glucocorticoids
Long-Term Relapse Prophylaxis (AQP4-Seropositive)
Four FDA-approved monoclonal antibody therapies exist for AQP4-seropositive NMOSD:
| Drug | Mechanism | Attack Risk Reduction | Notes |
|---|---|---|---|
| Eculizumab (Soliris) | Terminal complement inhibitor (anti-C5) | 94% (p<0.001) | Add-on to immunosuppression |
| Ravulizumab (Ultomiris) | Terminal complement inhibitor (anti-C5, longer-acting) | 100% (p<0.001) | Add-on to immunosuppression |
| Inebilizumab (Uplizna) | Anti-CD19 B-cell depleter | 78% (p=0.01) | Monotherapy; 300mg IV, then 2 weeks later, then every 6 months |
| Satralizumab (Enspryng) | IL-6 receptor blocker | 77% | Monotherapy; SC injection |
Preferred first-line approach (Harrison's 22e, 2025): Start with inebilizumab or satralizumab; use complement inhibitors (eculizumab/ravulizumab) as second-line for non-responders.
For AQP4-Seronegative NMOSD
No FDA-approved agents. Use empirical therapies:
- Rituximab (anti-CD20 B-cell depleter)
- Mycophenolate mofetil
- Azathioprine (older option)
Important warning: MS disease-modifying therapies (interferons, natalizumab, fingolimod) can worsen NMOSD and must be avoided.
Prognosis
- 85% have a relapsing course with severe exacerbations and poor recovery
- Disability accumulates from attack-related injury (not progressive course)
- AQP4-IgG-positive patients: more severe attacks, higher relapse rate (81-91%), worse outcomes
- Risk of respiratory failure and death from high cervical myelitis
- The most recent Bayesian network meta-analysis (John et al., J Neurol 2025) confirms that complement inhibitors (eculizumab/ravulizumab) offer the highest attack risk reduction in AQP4+ NMOSD among approved therapies
Sources:
- Harrison's Principles of Internal Medicine, 22nd ed. (2025), Chapter 456
- Bradley and Daroff's Neurology in Clinical Practice, Chapter 80
- Adams and Victor's Principles of Neurology, 12th Edition, Chapter 36
- Robbins, Cotran & Kumar Pathologic Basis of Disease
- Grainger & Allison's Diagnostic Radiology
- 2025 Brazilian NMOSD Guidelines (Apostolos-Pereira et al., Arq Neuropsiquiatr)
- MRI differentiation: NMOSD vs MS vs MOGAD (Tseriotis et al., JNNP 2025)
This is a shared conversation. Sign in to Orris to start your own chat.