TCA cycle
Reading File
Finding Sources
Searching the Web
TCA cycle steps enzymes NADH FADH2 regulation
Reading File
Reading File
Reading File
Reading File
Loading Image
Here is a thorough breakdown of the TCA cycle, drawn from Harper's Illustrated Biochemistry and Guyton & Hall Medical Physiology.
The TCA Cycle (Citric Acid / Krebs Cycle)
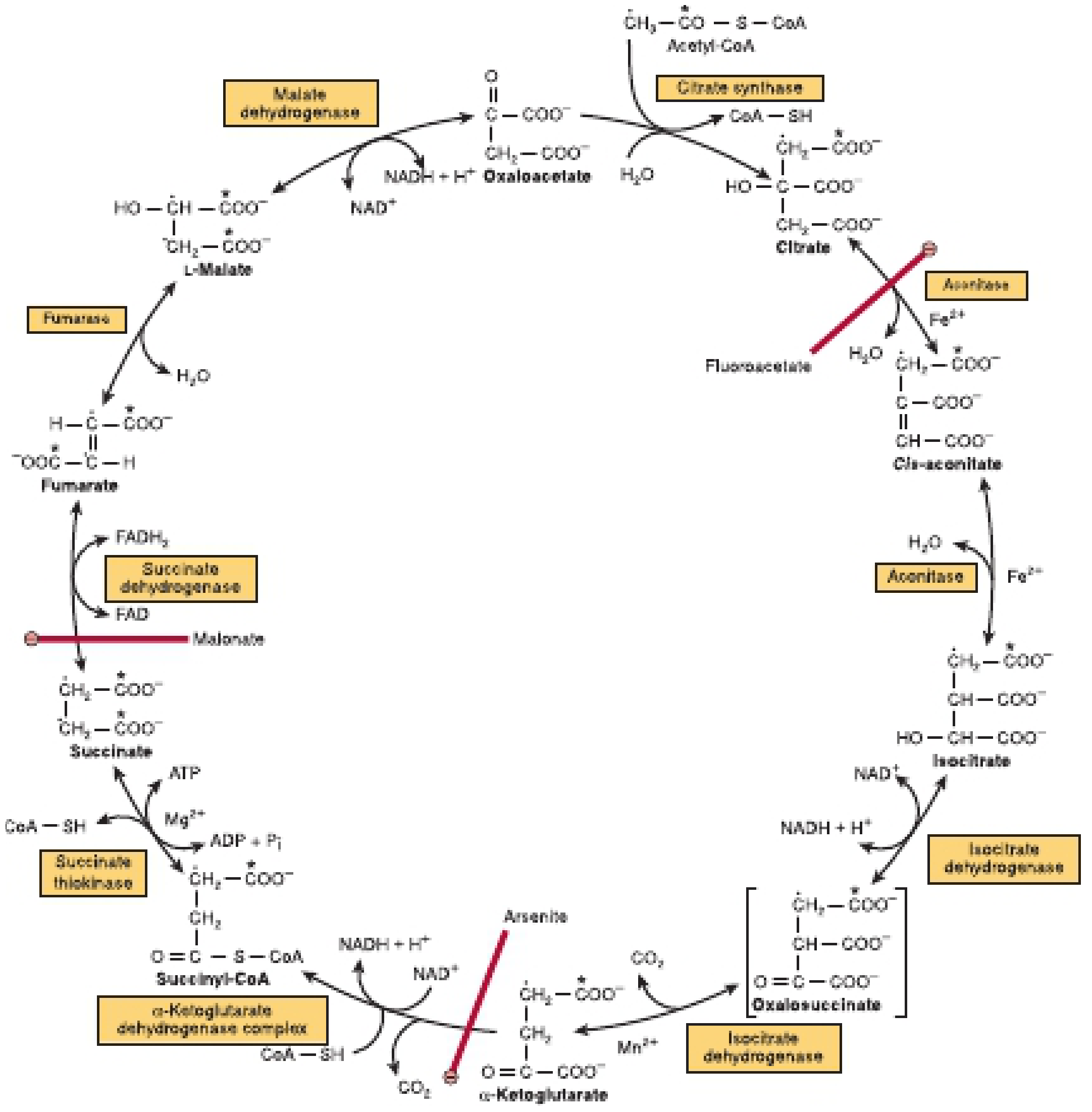
Location & Overview
- Occurs in the mitochondrial matrix (enzymes are free or bound to the inner mitochondrial membrane/cristae)
- Entry substrate: acetyl-CoA (2-carbon unit from carbohydrate, fat, and protein catabolism)
- Acetyl-CoA condenses with oxaloacetate (OAA, 4C) to form citrate (6C), then two CO2 are released and OAA is regenerated
- The cycle is amphibolic - serves both oxidative (energy) and biosynthetic (anabolic) functions
The 8 Reactions
| Step | Substrate → Product | Enzyme | Cofactor/Product |
|---|---|---|---|
| 1 | Acetyl-CoA + OAA → Citrate | Citrate synthase | CoA-SH released |
| 2 | Citrate → Isocitrate | Aconitase (via cis-aconitate) | Fe²⁺; H₂O removed then added |
| 3 | Isocitrate → α-Ketoglutarate + CO₂ | Isocitrate dehydrogenase | NADH produced; Mn²⁺ |
| 4 | α-Ketoglutarate → Succinyl-CoA + CO₂ | α-Ketoglutarate dehydrogenase complex | NADH produced; thiamine (B1), lipoate, FAD, CoA |
| 5 | Succinyl-CoA → Succinate | Succinate thiokinase | GTP (substrate-level phosphorylation); Mg²⁺ |
| 6 | Succinate → Fumarate | Succinate dehydrogenase | FADH₂ produced |
| 7 | Fumarate → Malate | Fumarase | H₂O added |
| 8 | Malate → Oxaloacetate | Malate dehydrogenase | NADH produced |
Steps 3 and 4 are the two CO₂-releasing steps.
Energy Yield Per Acetyl-CoA (One Turn)
| Product | Quantity | ATP equivalents |
|---|---|---|
| NADH | 3 | ~2.5 ATP each = 7.5 ATP |
| FADH₂ | 1 | ~1.5 ATP each = 1.5 ATP |
| GTP/ATP | 1 | 1 ATP (substrate-level) |
| Total | ~10 ATP |
Since one glucose yields 2 pyruvate → 2 acetyl-CoA, the TCA cycle contributes ~20 ATP per glucose (plus the 5 NADH from pyruvate dehydrogenase across two turns).
Vitamins Required
Four B vitamins are essential (Harper's, p. 171):
- Thiamine (B1) - thiamine diphosphate; cofactor for α-ketoglutarate dehydrogenase
- Riboflavin (B2) - FAD; cofactor for succinate dehydrogenase
- Niacin (B3) - NAD⁺; electron acceptor for isocitrate dehydrogenase, α-ketoglutarate dehydrogenase, and malate dehydrogenase
- Pantothenic acid (B5) - component of coenzyme A; needed to form acetyl-CoA and succinyl-CoA
Deficiency in any of these impairs TCA cycle function and energy metabolism.
Regulation
The three key regulatory enzymes are all inhibited by NADH and activated by ADP/NAD⁺ (reflecting energy status):
| Enzyme | Activated by | Inhibited by |
|---|---|---|
| Citrate synthase | Acetyl-CoA, OAA | NADH, citrate, succinyl-CoA, ATP |
| Isocitrate dehydrogenase | ADP, Ca²⁺, NAD⁺ | NADH, ATP |
| α-Ketoglutarate dehydrogenase complex | Ca²⁺, ADP | NADH, succinyl-CoA, ATP |
In high-energy states (high NADH, ATP), the cycle slows. In low-energy states (high ADP, NAD⁺), it accelerates.
Inhibitors (Toxicological/Experimental)
- Fluoroacetate (rat poison) - converted to fluorocitrate, inhibits aconitase (step 2)
- Malonate - competitive inhibitor of succinate dehydrogenase (step 6)
- Arsenite - inhibits the α-ketoglutarate dehydrogenase complex (step 4) by reacting with lipoate cofactor
Amphibolic Role: Beyond Energy
The TCA cycle also serves as a hub for biosynthesis (Harper's, p. 171):
- Gluconeogenesis - OAA → phosphoenolpyruvate via PEPCK
- Amino acid synthesis - transamination of α-ketoglutarate → glutamate; OAA → aspartate
- Fatty acid synthesis - citrate exported to cytoplasm → cleaved back to acetyl-CoA (by ATP-citrate lyase)
- Heme synthesis - succinyl-CoA condenses with glycine to form δ-aminolevulinic acid (ALA)
Anaplerosis and Cataplerosis
To sustain the cycle, carbon entering (anaplerosis) must equal carbon exiting (cataplerosis):
- Main anaplerotic reaction: pyruvate + CO₂ → OAA, catalyzed by pyruvate carboxylase (biotin-dependent; activated by acetyl-CoA)
- Other sources: glutamine/glutamate → α-ketoglutarate; propionyl-CoA → succinyl-CoA
- Cataplerosis: OAA → PEP (gluconeogenesis); citrate → cytoplasm (fatty acid synthesis)
Key Clinical Points
- Thiamine deficiency (beriberi, Wernicke's encephalopathy) - impairs α-ketoglutarate dehydrogenase, leading to accumulation of α-ketoglutarate and neurological dysfunction
- Pyruvate dehydrogenase deficiency - reduces acetyl-CoA supply to the cycle; lactic acidosis
- IDH1/IDH2 mutations (gliomas, AML) - mutant enzyme produces 2-hydroxyglutarate (oncometabolite) instead of α-ketoglutarate, disrupting epigenetic regulation
- Fumarate hydratase / succinate dehydrogenase mutations - tumor suppressors; loss causes accumulation of fumarate/succinate, inhibiting HIF prolyl hydroxylases → pseudo-hypoxia and tumor formation
Sources: Harper's Illustrated Biochemistry 32nd Ed, Chapter 16; Guyton & Hall Medical Physiology, Chapter 68
This is a shared conversation. Sign in to Orris to start your own chat.