Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Loading Image
I now have comprehensive content from three major textbooks and two key diagrams. Here is the full explanation:
Mechanism of Acute Respiratory Distress Syndrome (ARDS)
ARDS is a heterogeneous inflammatory syndrome - not a single disease - defined by acute onset, bilateral pulmonary opacities, severe hypoxemia (PaO2/FiO2 <300), and an absence of cardiogenic cause. The underlying lesion is diffuse alveolar damage (DAD), and the mechanism unfolds in three overlapping phases.

Precipitating Causes
ARDS is triggered by either direct (pulmonary) or indirect (extrapulmonary) insults:
- Direct: pneumonia, aspiration of gastric contents, inhalational injury, near-drowning
- Indirect: sepsis (most common overall cause), pancreatitis, massive blood transfusion (TRALI), trauma, burns
In pancreatitis, for example, pancreatic elastase and lipase increase vascular permeability, phospholipase A2 degrades surfactant, and systemically released TNF-α and IL-8 drive neutrophil sequestration in the lungs. - Murray & Nadel's Textbook of Respiratory Medicine
Phase 1: Exudative Phase (Days 0-7)
This is the core mechanism of ARDS.
1. Alveolar-Capillary Barrier Disruption
The alveolar wall is formed by two cell layers - the capillary endothelium and the alveolar epithelium (type I and type II pneumocytes) - linked by tight junctions. In health, the epithelium's tight junctions prevent fluid from leaking into the air spaces, and active ion transport (Na+/K+-ATPase and ENaC channels in type II cells) continuously clears fluid from the alveolar space.
In ARDS, an inflammatory insult disrupts both barriers by:
- Disrupting intercellular tight junctions
- Inducing death and sloughing of endothelial and epithelial cells
This causes a massive increase in alveolar-capillary permeability and the influx of protein-rich edema fluid into the alveolar and interstitial spaces. - Murray & Nadel's, p. 187
2. Cytokine Storm and Neutrophil Recruitment
The initial insult (bacteria, viruses, or sterile injury) activates Toll-like receptors on alveolar type I cells and resident alveolar macrophages, triggering release of proinflammatory chemokines and cytokines: IL-1, IL-6, IL-8, TNF-α, leukotriene B4. These recruit neutrophils (PMNs) from the circulation into the pulmonary interstitium and alveoli. - Harrison's Principles of Internal Medicine 22E
3. Neutrophil-Mediated Injury
Neutrophils are the primary early effectors of DAD. Recruited neutrophils release:
- Neutrophil elastase and serine proteases: Elevated in BAL fluid; correlate with injury severity; degrade junction proteins in both epithelium and endothelium
- Matrix metalloproteinases (MMPs): Released by both neutrophils and macrophages; degrade junctional proteins and induce cell death
- Reactive oxygen species (ROS) and reactive nitrogen species: Overwhelm the lung's antioxidant defenses; cause epithelial and endothelial cell death, disrupt tight junctions, and potentiate proteinase activity by inactivating antiproteinases
- Neutrophil extracellular traps (NETs): Cause direct epithelial and endothelial injury
Importantly, ARDS can develop even in neutropenic patients, confirming that neutrophils are not the sole mediators. - Murray & Nadel's, p. 187
4. Monocytes, Macrophages, and Platelets
Monocytes migrate into the lung after the initial neutrophilic wave and acquire a macrophage phenotype, secreting cytokines, chemokines, MMPs, TNF-α, TRAIL (TNF-related apoptosis-inducing ligand via IFN-β), and VEGF - all of which propagate injury. Platelet-PMN aggregates further increase vascular permeability. Free hemoglobin from red blood cells that extravasate into the airspace exacerbates injury via oxidant mechanisms. - Murray & Nadel's, p. 187
5. Consequences
The alveolar flooding and cellular damage produce:
- Hyaline membrane formation: Condensed plasma proteins, cellular debris, and dysfunctional surfactant aggregate to form characteristic whorls lining the denuded alveolar walls
- Surfactant inactivation: Direct enzymatic destruction (e.g., by phospholipase A2) and dilution by edema fluid; decreased production by injured type II cells - further reducing compliance and promoting alveolar collapse
- Intrapulmonary shunting: Edema floods dependent alveoli (shown on CT as dense, gravity-dependent consolidation), while ventilation continues to non-dependent zones - creating V/Q mismatch and right-to-left shunt - the cause of severe, refractory hypoxemia
- Pulmonary vascular injury: Microthrombi and fibrocellular proliferation obliterate microvasculature, increasing dead space and pulmonary vascular resistance, contributing to pulmonary hypertension and CO2 retention (hypercapnia)
- Impaired alveolar fluid clearance: Hypoxia and hypercapnia themselves inhibit Na+/K+-ATPase and ENaC, further reducing the lung's ability to clear edema - Harrison's, p. 2343-2344
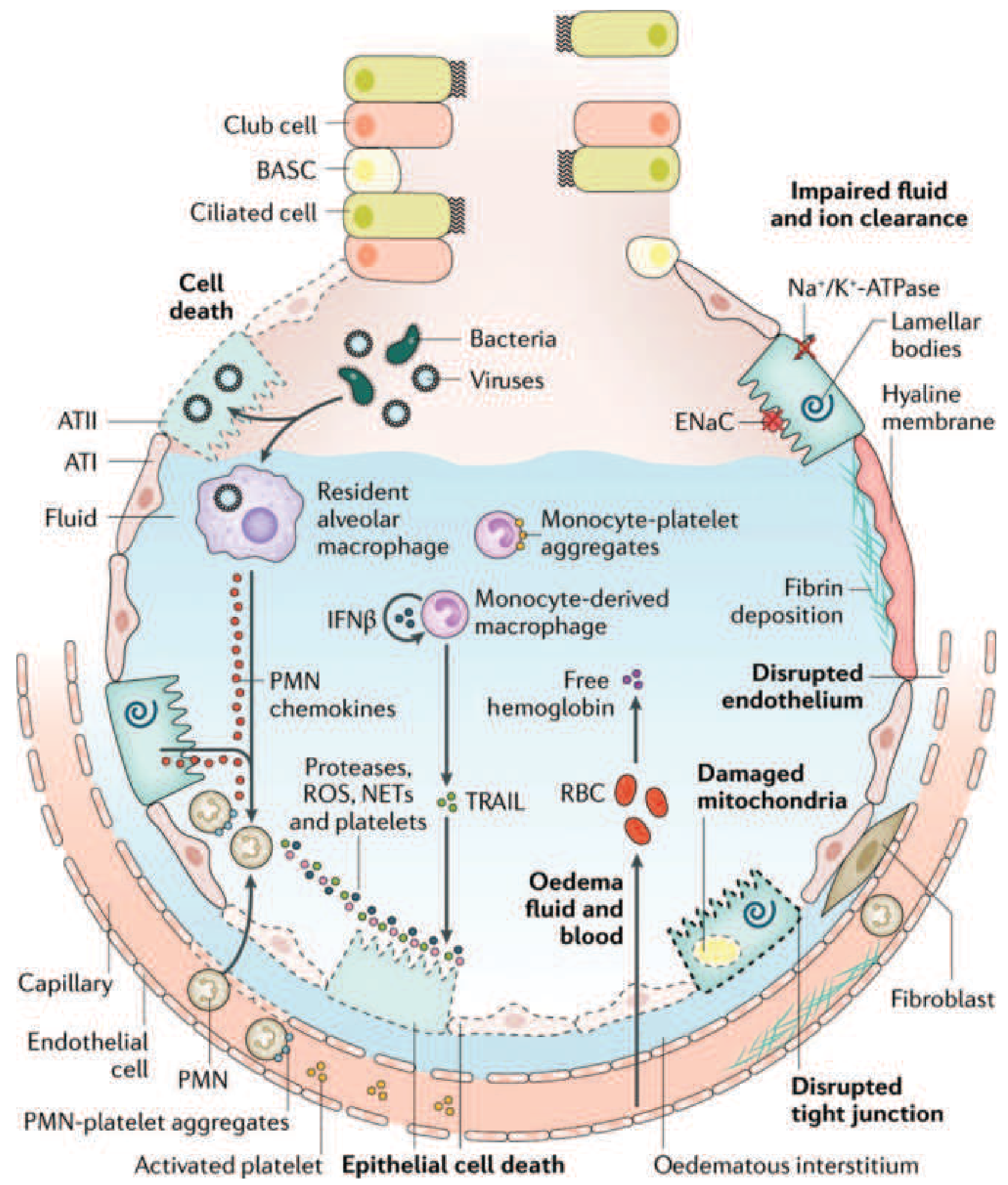
Figure: The injured alveolus - Harrison's Principles of Internal Medicine 22E (after Matthay et al., Nat Rev Dis Primers, 2019)
Phase 2: Proliferative Phase (Days 7-14)
By day 7, a reparative response begins alongside ongoing injury:
- Infiltrate shifts from neutrophil-predominant to lymphocyte-predominant
- Type II pneumocytes proliferate along denuded alveolar basement membranes, synthesizing new surfactant and differentiating into type I pneumocytes to restore the epithelial barrier
- Alveolar exudates begin to organize
- In many patients, lung function gradually recovers over 3-4 weeks - Harrison's, p. 2344
Phase 3: Fibrotic Phase (Day 21+, subset of patients)
Some patients fail to resolve and enter a fibrotic phase:
- Alveolar-duct and interstitial fibrosis replaces the exudative and inflammatory material
- Marked acinar architectural disruption leads to emphysema-like changes with large bullae
- Intimal fibroproliferation in the pulmonary microcirculation causes progressive vascular occlusion and sustained pulmonary hypertension
- Clinically: risk of pneumothorax, reduced compliance, increased dead space, prolonged ventilator dependence
- Lung biopsy evidence of fibrosis in any ARDS phase predicts increased mortality - Harrison's, p. 2344
Ventilator-Induced Lung Injury (VILI): A Secondary Mechanism
Mechanical ventilation itself can perpetuate lung injury via two mechanisms:
- Volutrauma/Barotrauma: High tidal volumes overdistend "baby lung" regions (the relatively normal, non-dependent zones that receive most of the ventilation in ARDS), triggering further inflammatory injury - indistinguishable histologically from ARDS itself
- Atelectrauma: Repeated opening and collapse of unstable alveoli at end-expiration, amplifying shear stress injury
This is why lung-protective ventilation (6 mL/kg predicted body weight, plateau pressure ≤30 cmH2O) reduces ARDS mortality by ~9% in the landmark ARDSNet trial. - Harrison's, p. 2345; Goldman-Cecil Medicine
Summary Table
| Feature | Mechanism |
|---|---|
| Non-cardiogenic edema | Barrier disruption (endothelial + epithelial tight junctions) |
| Refractory hypoxemia | Intrapulmonary shunting (flooded alveoli, V/Q mismatch) |
| Decreased compliance | Surfactant loss + alveolar flooding + atelectasis |
| Hypercapnia | Increased dead space (microvascular obliteration) |
| Hyaline membranes | Fibrin + protein + cellular debris deposition |
| Pulmonary hypertension | Microvascular obliteration + vasoconstriction |
| Bilateral opacities | Gravity-dependent edema + inflammation |
Sources: Murray & Nadel's Textbook of Respiratory Medicine; Harrison's Principles of Internal Medicine 22E; Fishman's Pulmonary Diseases and Disorders; Goldman-Cecil Medicine.
This is a shared conversation. Sign in to Orris to start your own chat.