
The patient with mental retardness with these type of overgrowths on face. What’s this condition?
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
The clinical picture is classic. Here's the diagnosis and a full breakdown:
Tuberous Sclerosis Complex (TSC)
The facial lesions visible in this patient — multiple small, firm, dome-shaped papules and nodules distributed symmetrically over the cheeks, nose, and nasolabial folds — are facial angiofibromas, previously misnamed "adenoma sebaceum." Combined with intellectual disability (mental retardation), this is the hallmark presentation of Tuberous Sclerosis Complex.
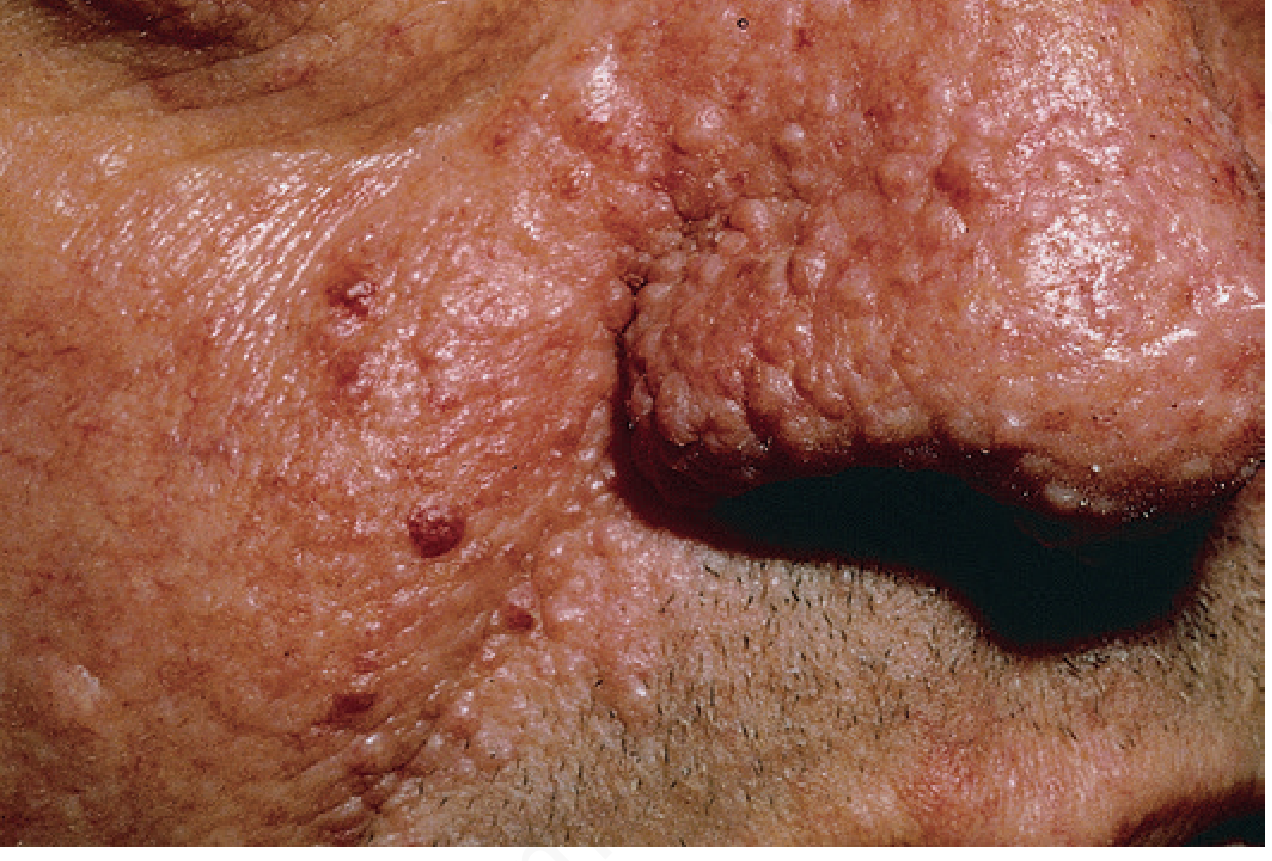
Angiofibromas (adenoma sebaceum) — Andrews' Diseases of the Skin, Fig. 27.9
What is TSC?
TSC is an autosomal dominant neurocutaneous syndrome (phakomatosis) caused by mutations in TSC1 (hamartin) or TSC2 (tuberin), which are tumor suppressor genes regulating the mTOR signaling pathway. Loss-of-function leads to uncontrolled cell proliferation and hamartoma formation across multiple organ systems.
Classic Triad (Vogt's Triad)
- Facial angiofibromas (adenoma sebaceum)
- Intellectual disability / mental retardation
- Epilepsy / seizures
(Present together in only a minority of patients, but facial angiofibromas + intellectual disability as seen here strongly suggest TSC)
Cutaneous Features of TSC
| Feature | Description |
|---|---|
| Facial angiofibromas | 1–3 mm yellowish-red waxy papules over cheeks, nose, forehead; present in ~90% of patients >4 yrs |
| Ash-leaf macules | Hypopigmented oval/leaf-shaped patches; earliest cutaneous sign (85% of patients) |
| Shagreen plaque | Roughened, skin-colored collagenoma typically on lower back |
| Periungual/subungual fibromas | Flesh-colored growths emerging from nail folds |
| Fibrous cephalic plaque | Firm fibrous plaque on forehead/scalp |
Systemic Involvement
- Brain: Cortical tubers, subependymal nodules, subependymal giant cell astrocytomas → seizures (80%), learning disability (>50%), autism spectrum disorder
- Kidney: Angiomyolipomas (80%), renal cysts, rarely renal cell carcinoma
- Heart: Rhabdomyomas (especially in infancy, usually regress)
- Lung: Lymphangioleiomyomatosis (mainly in women)
- Eye: Retinal hamartomas, achromic patches
Management
- mTOR inhibitors (everolimus, sirolimus/rapamycin) — systemic for CNS/renal lesions and topical rapamycin for facial angiofibromas
- Antiepileptics for seizure control
- Laser therapy or dermabrasion for cosmetic treatment of angiofibromas
- Regular surveillance: MRI brain, renal ultrasound, echocardiogram, pulmonary function
Bottom line: This patient's combination of diffuse facial angiofibromas (the clustered papules/nodules in a butterfly distribution) and intellectual disability is pathognomonic for Tuberous Sclerosis Complex. Genetic testing for TSC1/TSC2 mutations and multi-organ evaluation are warranted.
— Andrews' Diseases of the Skin, p. 643; Emery's Elements of Medical Genetics and Genomics, p. 308
Tuberous Sclerosis Complex
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Here is a comprehensive overview of Tuberous Sclerosis Complex from the authoritative textbooks:
Tuberous Sclerosis Complex (TSC)
History & Overview
First described by Désiré-Magloire Bourneville (1880), who linked cerebral lesions to the facial skin changes. Vogt (1890) formally delineated the classic triad. The condition is also called Bourneville Disease or Epiloia (Sherlock, 1911 — term never widely adopted).
TSC is a neurocutaneous syndrome (phakomatosis) characterized by hamartomatous proliferations in multiple organs, arising from a fundamental defect in tumor suppressor function.
Genetics & Pathogenesis
| Feature | Detail |
|---|---|
| Inheritance | Autosomal dominant, variable penetrance |
| Gene 1 | TSC1 — chromosome 9q → encodes Hamartin |
| Gene 2 | TSC2 — chromosome 16p → encodes Tuberin |
| Mechanism | TSC2 is adjacent to PKD1; some deletions affect both genes simultaneously |
| Protein function | Hamartin + tuberin interact physically to suppress mTOR signaling and inhibit cell growth |
| Loss of heterozygosity | Both alleles must be affected for full disease expression ("two-hit" mechanism) |
| Spontaneous mutations | Up to 50% of cases; ~15% of sporadic cases show no identifiable mutation (likely mosaicism) |
| Prevalence | 1 in 5,800–15,000 births |
Key molecular mechanism: Hamartin and tuberin normally inhibit the mTOR (mammalian target of rapamycin) pathway. Mutations cause uncontrolled mTOR activation → abnormal cell growth, proliferation, and hamartoma formation across ectodermal and mesodermal tissues. Giant neurons (3–4× normal size) are seen in cerebral tubers due to this dysregulated cell-size pathway.
Classic Triad (Vogt's Triad)
- Facial angiofibromas (adenoma sebaceum)
- Epilepsy / seizures
- Intellectual disability / developmental delay
The complete triad is present in only a minority of patients. Many have forme fruste (incomplete) presentations.
Clinical Features
🔴 Cutaneous Manifestations
| Lesion | Description | Frequency |
|---|---|---|
| Facial angiofibromas (adenoma sebaceum) | 1–3 mm yellowish-red, waxy, dome-shaped papules; symmetric over cheeks, nose, nasolabial folds, chin, forehead. Earliest sign may be mild erythema over cheeks. NOT true adenomas — actually angiofibromas. | 90% of patients >4 yrs |
| Ash-leaf macules (hypomelanotic macules) | Oval, leaf-shaped white patches; first cutaneous lesion to appear; congenital or develops by 6–8 yrs; enhanced by Wood's lamp (UV 360 nm) | 85–90% |
| Shagreen patch | Roughened, skin-colored plaque of subepidermal fibrosis; typically on lower back/trunk | Common |
| Periungual/subungual fibromas (Koenen tumors) | Small, digitate, protruding fibromas at nail folds; appear at puberty | ~50% |
| Fibrous cephalic plaque | Firm fibrous plaque on forehead/scalp; marker for severe intracranial involvement | Present |
| Café-au-lait spots, confetti macules, poliosis | Additional cutaneous markers | Variable |
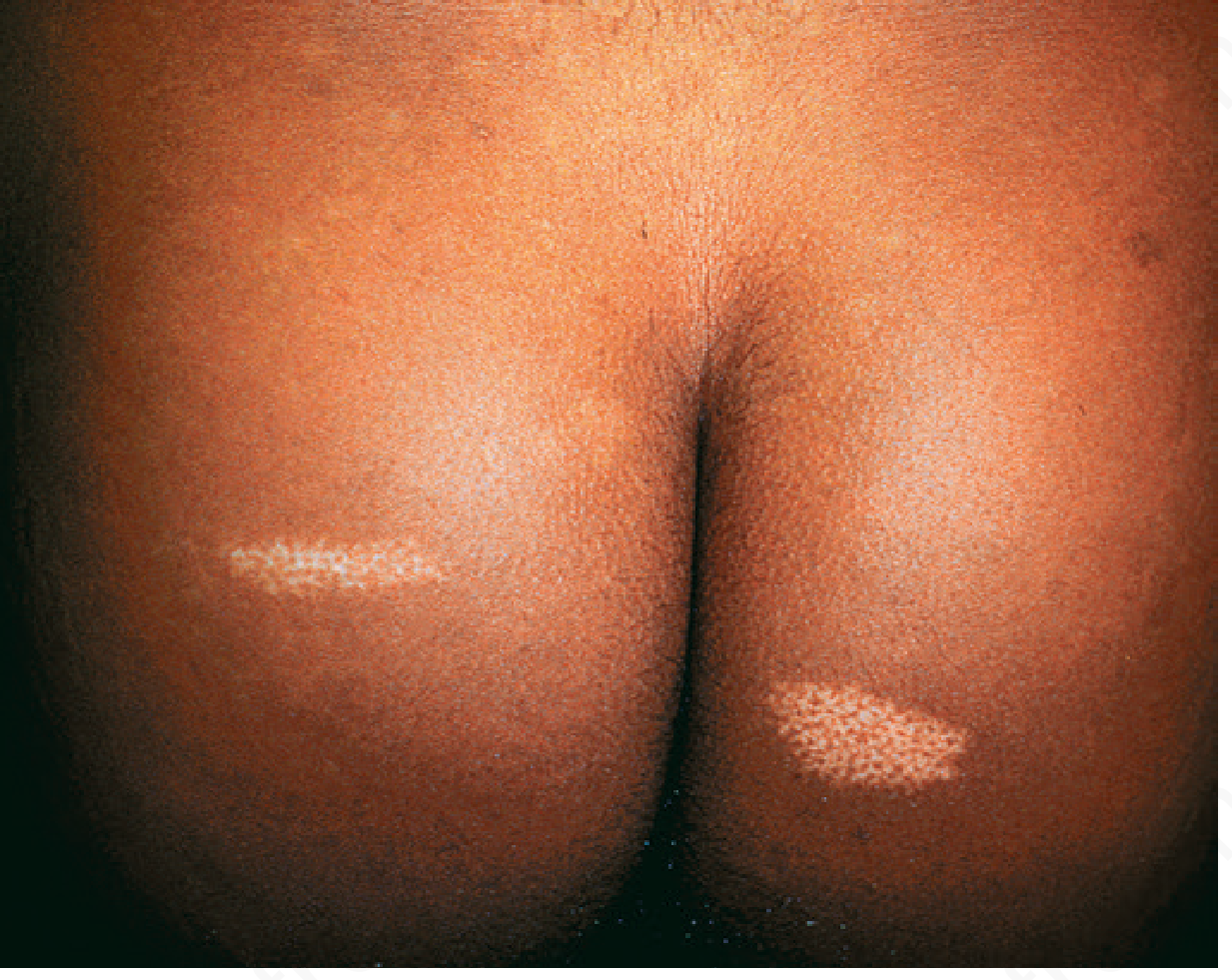
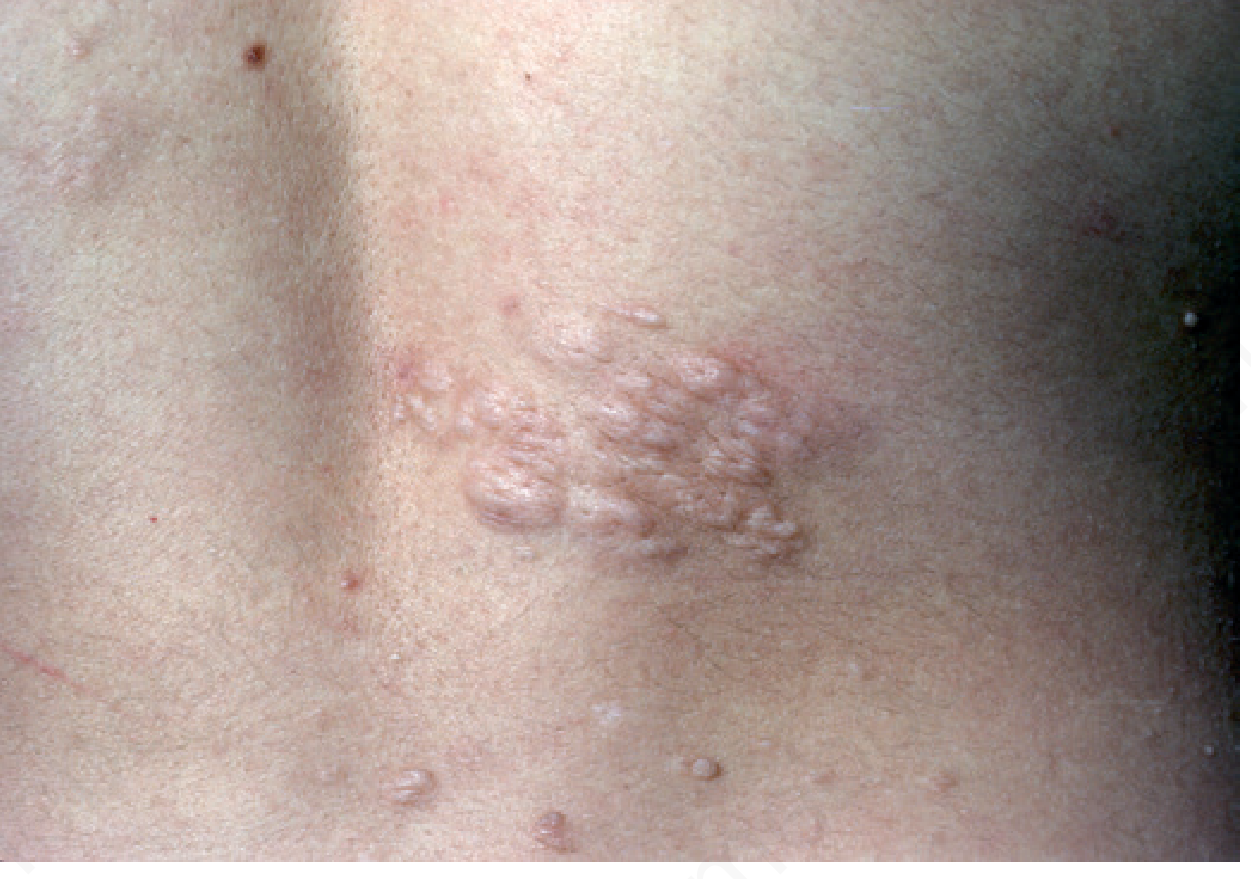
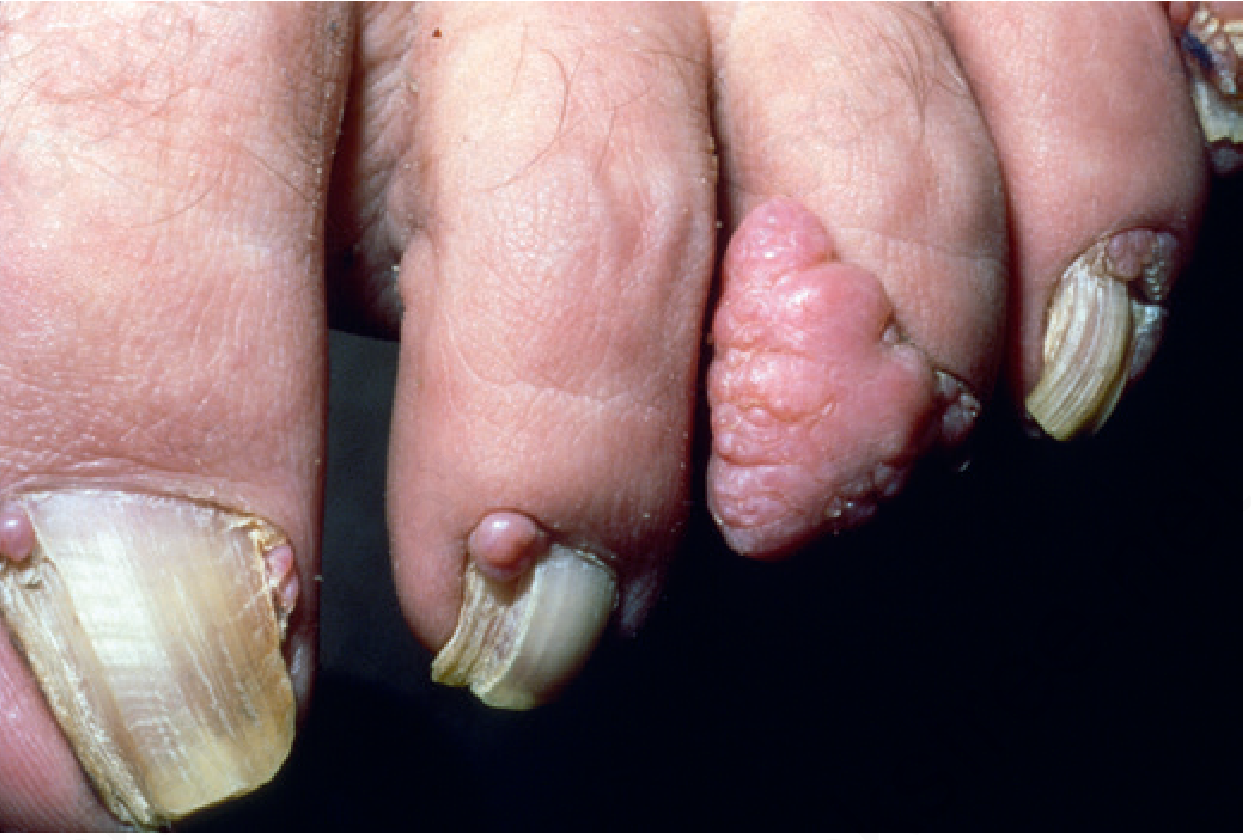
🧠 Neurological Manifestations
- Seizures: 80–90% of patients. Initial pattern = infantile spasms with hypsarrhythmia (25% of all infantile spasms are TSC). Progress over years to generalized motor, psychomotor, or atypical absence attacks
- Cortical tubers: Potato-like hamartomatous nodules in the cortex; calcify over time (visible on CT); visible on MRI as early as 6 weeks of age
- Subependymal nodules: "Candle drippings" along ventricular walls
- Subependymal giant cell astrocytomas (SEGA): May progress from subependymal nodules
- Intellectual disability: 40–60% of patients; severity varies widely. Early seizure onset is predictive of developmental delay
- Autism spectrum disorder / ADHD / behavioral disturbances: Common neurodevelopmental comorbidities
🫀 Systemic Organ Involvement
| Organ | Manifestation | Notes |
|---|---|---|
| Kidney | Angiomyolipomas (bilateral, multiple) — most common renal finding; renal cysts (20–30%); rarely renal cell carcinoma | AMLs >4 cm → prophylactic surgery; regular imaging screening for RCC |
| Heart | Rhabdomyomas (43%) | Highly specific for TSC when found on fetal echo; usually regress after birth; may cause conduction defects |
| Lung | Lymphangioleiomyomatosis (LAM) | Predominantly women in 30s–40s; progressive respiratory failure, pneumothorax; smooth muscle proliferation + cystic degeneration |
| Eye | Retinal hamartomas (phakomas) — gray/yellow plaques near optic disc; achromic patches; nystagmus, angioid streaks | ~50%; van der Hoeve coined "phakomatosis" from these lesions |
| Liver, thyroid, testes, GI | Angiomyolipomas | Less common |
| Bone | Bone cysts (digits), sclerotic lesions, "marbling" | ~50% |
| Teeth | ≥5 dental enamel pits in permanent teeth | Diagnostic marker |
| Oral | Gingival fibromas, buccal/labial/lingual papillomatosis | Present |
Diagnostic Criteria (2012 International TSC Consensus Conference)
Definite Diagnosis: 2 major criteria OR 1 major + ≥2 minor criteria
Possible Diagnosis: 1 major OR ≥2 minor criteria
Genetic: Pathogenic mutation in TSC1 or TSC2 alone is sufficient
Major Criteria
- ≥3 hypomelanotic macules (≥5 mm)
- ≥3 angiofibromas OR fibrous cephalic plaque
- ≥2 periungual fibromas
- Shagreen patch
- Retinal hamartomas
- Cortical dysplasia (tubers)
- Subependymal nodules
- Subependymal giant cell astrocytoma (SEGA)
- Cardiac rhabdomyoma
- Lymphangioleiomyomatosis
- Angiomyolipomas (≥2)
Minor Criteria
- Confetti-like skin macules
- ≥3 dental enamel pits
- ≥2 intraoral fibromas
- Retinal achromic patch
- Multiple renal cysts
- Nonrenal hamartomas
Workup / Investigations
| Investigation | Purpose |
|---|---|
| Wood's lamp exam | Detect subtle ash-leaf macules |
| MRI brain | Detect cortical tubers, subependymal nodules, SEGA (detectable as early as 6 weeks) |
| CT brain | Calcified intracranial nodules in older patients |
| Renal ultrasound / MRI | Angiomyolipomas, cysts, RCC screening |
| Echocardiogram | Cardiac rhabdomyomas (especially in infants/fetus) |
| Funduscopy | Retinal phakomas |
| EEG | Characterize seizure type; hypsarrhythmia in infancy |
| Genetic testing | TSC1/TSC2 mutation analysis |
| Pulmonary function / HRCT chest | LAM screening in women |
| Skin biopsy | Histopathology of angiofibromas if needed |
Treatment
Pharmacological
| Agent | Use |
|---|---|
| Everolimus (mTOR inhibitor) | First-line for SEGAs, renal AMLs, epilepsy; FDA/EMA approved |
| Sirolimus (rapamycin) | Renal AMLs, LAM, facial angiofibromas |
| Topical rapamycin (0.1–1%) | Non-invasive treatment of facial angiofibromas; lesions recur without maintenance |
| Antiepileptics | Seizure control; vigabatrin is particularly effective for infantile spasms in TSC |
Non-pharmacological
| Treatment | Indication |
|---|---|
| Laser therapy / dermabrasion / shaving | Cosmetic treatment of angiofibromas |
| Surgical resection | AMLs >4 cm (hemorrhage risk); nephron-sparing where possible |
| Avoid cranial irradiation of astrocytomas | Risk of subsequent glioblastoma development |
Key Points to Remember
- TSC2 mutations are more common than TSC1 and tend to cause more severe phenotype
- The severity of epilepsy, cognitive deficit, and skin lesions do NOT correlate with each other — a patient may have florid skin lesions with mild cognitive function, or vice versa
- Early seizure onset is the strongest predictor of intellectual disability
- Cardiac rhabdomyomas detected on fetal ultrasound are an important early marker for TSC
- mTOR inhibitors are now considered first-line systemic therapy
- TSC2 is adjacent to PKD1 — contiguous gene deletions cause combined TSC + ADPKD (polycystic kidney disease) phenotype
— Andrews' Diseases of the Skin, pp. 642–644 | Adams and Victor's Principles of Neurology, 12th Ed., pp. 1025–1026 | Harrison's Principles of Internal Medicine, 22nd Ed.
What radiological investigations are to be done in this case?
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Here is a complete, guideline-based radiology workup for a confirmed TSC patient:
Radiological Investigations in Tuberous Sclerosis Complex
Based on the 2021 International TSC Consensus Guidelines (Northrup et al., Pediatric Neurology), the radiological workup covers all potentially involved organ systems.
1. 🧠 Brain MRI — HIGHEST PRIORITY
Modality of choice: MRI Brain with and without gadolinium contrast
What to look for:
| Finding | MRI Appearance |
|---|---|
| Cortical tubers | Hypointense on T1, hyperintense on T2/FLAIR; expand gyri; may calcify |
| Subependymal nodules (SEN) | "Candle drippings" along ventricular walls; calcify over time → hypointense on T2 |
| Subependymal giant cell astrocytoma (SEGA) | Enhancing mass at foramen of Monro; >1 cm, growing → urgent treatment |
| White matter radial migration lines | T2-hyperintense lines radiating from ventricle to cortex |
| Hydrocephalus | May occur if SEGA obstructs CSF flow |
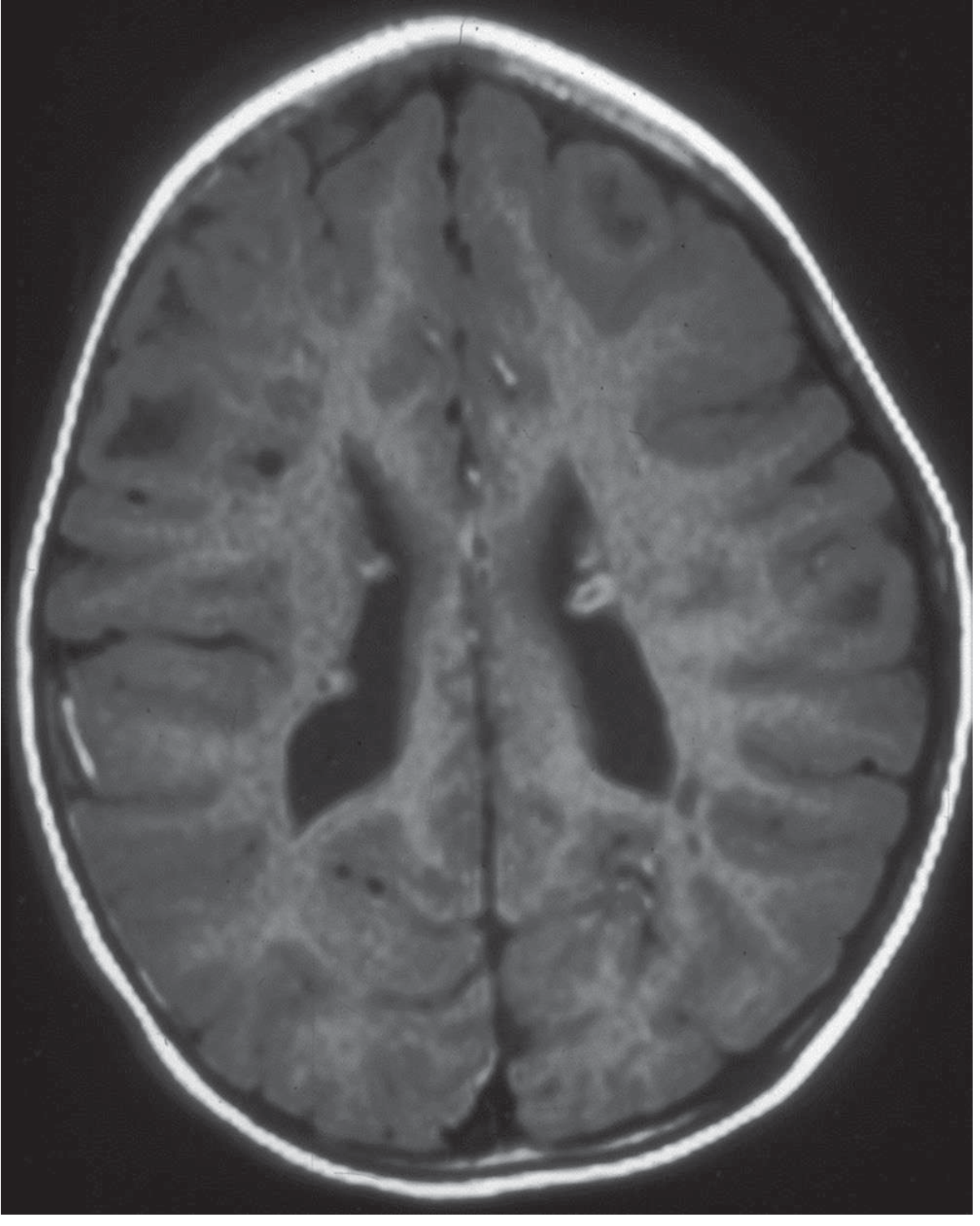
Note: CT or cranial ultrasound (in infants with open fontanelles) is suboptimal but acceptable if MRI is not available. CT detects calcified nodules well but misses early/uncalcified tubers. MRI detects lesions as early as 6 weeks of age.
Surveillance:
- MRI every 1–3 years until age 25 to monitor for SEGA growth
- More frequent if SEGA is large, growing, or symptomatic
- After age 25: as clinically indicated
2. 🫘 Abdominal MRI — Preferred over CT/Ultrasound
Modality of choice: MRI Abdomen (avoids radiation; better soft tissue characterization)
What to look for:
| Finding | Notes |
|---|---|
| Renal angiomyolipomas (AML) | Bilateral, multiple; fat-containing lesions; characteristic fat signal on MRI/CT |
| Renal cysts | Present in 20–30%; may mimic ADPKD if TSC2/PKD1 contiguous deletion |
| Renal cell carcinoma | Increased risk; screening mandatory |
| Hepatic AMLs | Less common |
Angiomyolipomas >3–4 cm → at risk for spontaneous hemorrhage (Wunderlich syndrome) → mTOR inhibitors or embolization
Surveillance:
- Every 1–3 years if no known renal lesions
- Annually if renal lesions present
- May alternate MRI with ultrasound or low-dose CT
3. 🫁 High-Resolution CT (HRCT) Chest
Indication: All women ≥18 years; adolescents and men if symptomatic
What to look for:
| Finding | Description |
|---|---|
| Lymphangioleiomyomatosis (LAM) | Diffuse bilateral thin-walled cysts throughout lung parenchyma |
| Multifocal micronodular pneumocyte hyperplasia | Bilateral nodules; can coexist with LAM |
Surveillance:
- Every 5–10 years until menopause in women without known LAM
- Annual pulmonary function tests (PFTs) if LAM is confirmed
4. 🫀 Echocardiography
Indication: All children (especially <3 years); symptomatic adults
What to look for:
| Finding | Notes |
|---|---|
| Cardiac rhabdomyomas | Echogenic intramural masses; highly specific for TSC when multiple; usually in ventricles |
| Obstruction / conduction defects | If atrial location |
Cardiac rhabdomyomas typically regress spontaneously after birth — do NOT require surgery unless causing obstruction or arrhythmia.
Surveillance:
- Every 1–3 years in children with rhabdomyomas until regression documented
- Every 3–5 years ECG in asymptomatic patients
5. 🦴 Skeletal X-rays (Hands, Feet, Spine)
- Bone cysts in digits (phalanges)
- Sclerotic lesions / "marbling" of bones
- Present in ~50% of patients
- Not routinely performed unless symptomatic; may be done as part of initial workup
6. 🦷 Dental Panoramic Radiograph
- By age 7 if dental asymmetry, swelling, or abnormal tooth development
- Detects dental enamel pits (minor diagnostic criterion) and jaw lesions
Summary Table — Radiology Workup at a Glance
| Investigation | Organ | Priority | Frequency |
|---|---|---|---|
| MRI Brain ± gadolinium | Brain | ⭐ Highest | Every 1–3 yrs till age 25 |
| MRI Abdomen | Kidney, Liver | ⭐ High | Every 1–3 yrs (annual if lesions) |
| HRCT Chest | Lung (LAM) | Women ≥18 yrs | Every 5–10 yrs |
| Echocardiography | Heart | Children <3 yrs | Every 1–3 yrs |
| ECG | Heart | All | Every 3–5 yrs |
| X-ray hands/feet | Bone | Symptomatic | As needed |
| Dental panoramic X-ray | Teeth/jaw | Age 7 if abnormal | Every 6–12 months |
Practical Note for This Patient
Given this patient's intellectual disability + facial angiofibromas, priority investigations are:
- MRI Brain with gadolinium — assess cortical tubers, SEGA, subependymal nodules (directly explains her cognitive impairment and seizure risk)
- Abdominal MRI/Ultrasound — screen for renal AMLs and cysts
- Echocardiography — rule out cardiac rhabdomyomas
- HRCT Chest — if she is ≥18 years or symptomatic
- EEG — not radiological, but essential to characterize seizure activity
— Dermatology 2-Volume Set 5e (Table 61.8); Goldman-Cecil Medicine, 22nd Ed.; Harrison's Principles of Internal Medicine, 22nd Ed.
This is a shared conversation. Sign in to Orris to start your own chat.