Familial adenomatous polyposis pathology
Reading File
Finding Sources
Finding Sources
Searching PubMed
"familial adenomatous polyposis"[MeSH Terms] AND pathology
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
I now have comprehensive material. Let me compile the full answer.
Familial Adenomatous Polyposis (FAP) - Pathology
Definition and Genetics
FAP is an autosomal dominant disorder caused by germline mutations of the APC (adenomatous polyposis coli) gene on chromosome 5q21-q22. It has 80-100% penetrance and an estimated prevalence of 1 in 5,000-7,500. About 75% of cases are inherited; the rest arise from de novo mutations.
The APC gene is a classic tumor suppressor gene - the mutated allele is inherited from the affected parent, and adenomas develop when the second (wild-type) allele is subsequently lost or mutated (two-hit model). Nearly all APC mutations create a premature stop codon, producing a truncated, non-functional protein.
- Robbins, Cotran & Kumar Pathologic Basis of Disease - Robbins & Kumar Basic Pathology, p. 584
Molecular Pathogenesis: The APC/Wnt Pathway
The APC protein is a key negative regulator of the Wnt signaling pathway:
- In normal cells, APC forms a destruction complex with axin, conductin, and GSK3-beta kinase
- This complex binds and phosphorylates beta-catenin, marking it for ubiquitin-mediated degradation in the cytoplasm
- When APC is mutated/truncated, beta-catenin is no longer degraded and accumulates in the cytoplasm
- Beta-catenin translocates to the nucleus and interacts with transcription factor TCF-4, upregulating genes that drive adenoma formation
- Truncated APC also disrupts normal chromosomal segregation, generating chromosomal instability (CIN)
Germline mutations are distributed throughout the 5' half of the APC gene; somatic mutations cluster in the central mutation cluster region (MCR) near codon 1309.
- Sleisenger and Fordtran's Gastrointestinal and Liver Disease, p. 2578
Gross and Histologic Pathology
Classic FAP requires at least 100 adenomatous polyps for diagnosis; thousands may be present (Fig. 17.51 from Robbins Cotran below).
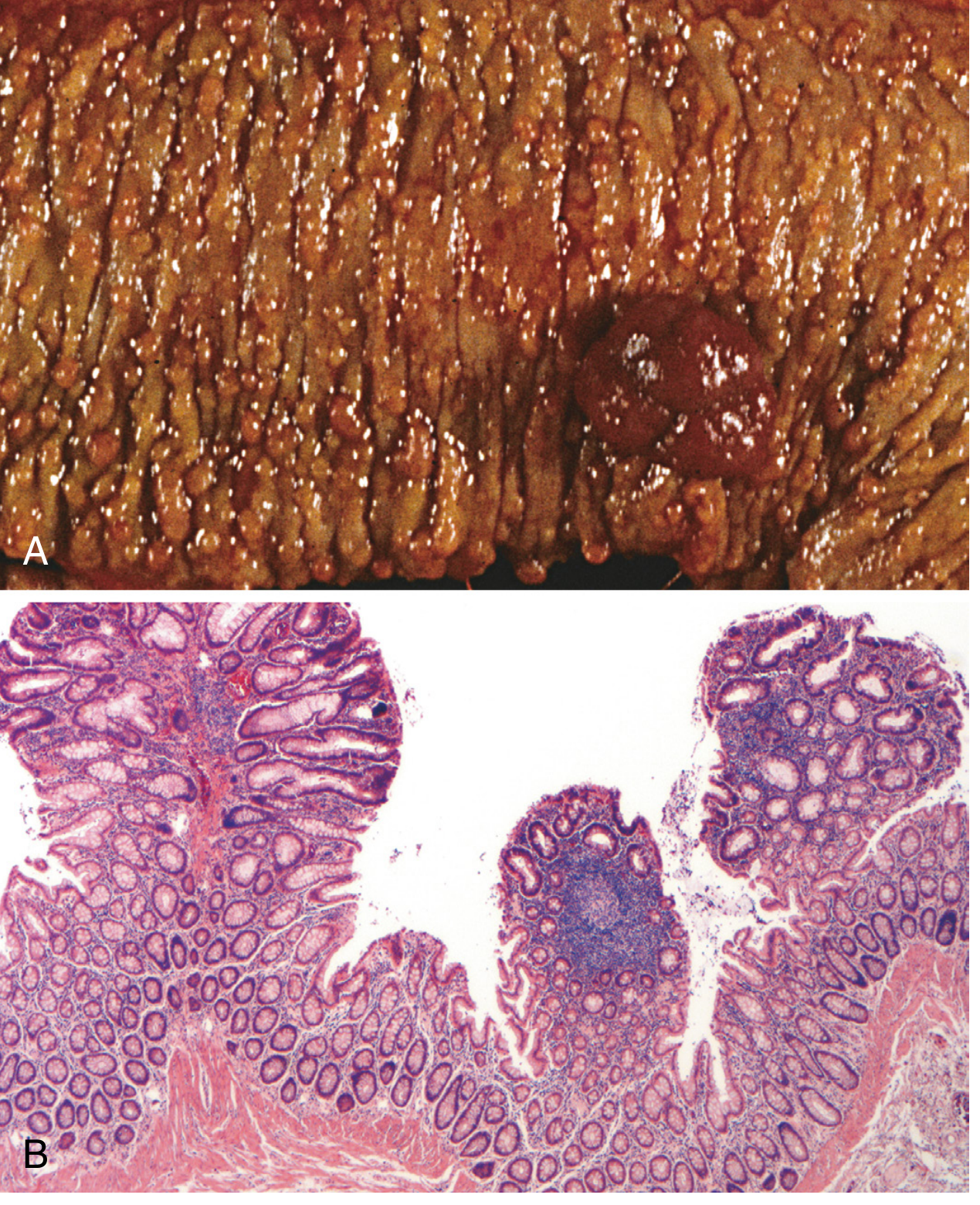
Key histologic features:
- FAP-associated polyps are morphologically indistinguishable from sporadic adenomas - tubular or villous architecture, dysplastic epithelium
- Flat or depressed adenomas are prevalent in FAP
- Microscopic adenomas (1-2 dysplastic crypts only) occur in otherwise normal-appearing mucosa
- Polyposis involves the entire colon and rectum with no predilection for a single region
The adenoma morphology below shows (A) pedunculated adenoma endoscopically, (B) velvety surface, and (C) tubular adenoma histology:
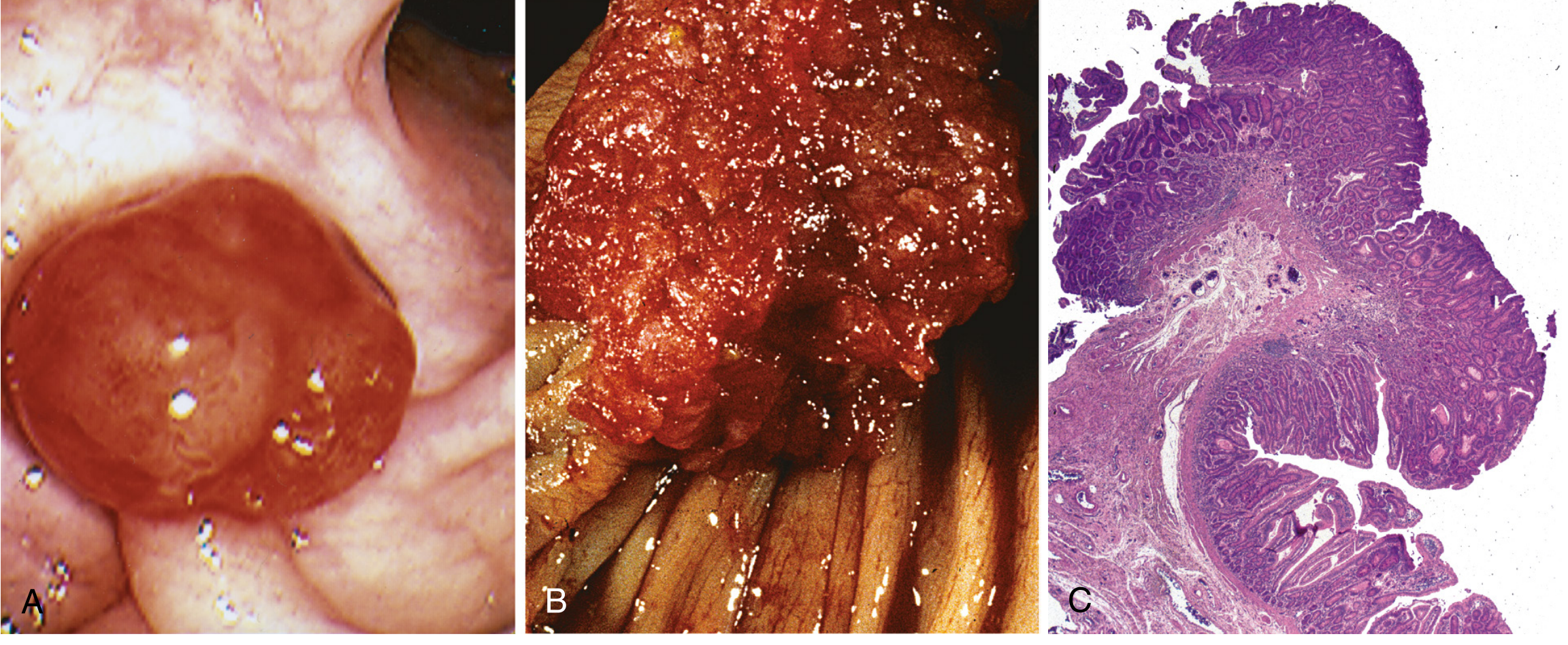
Malignant Potential
Colorectal adenocarcinoma develops in 100% of untreated FAP patients, often before age 30, nearly always by age 50. This is the cornerstone of the condition's clinical significance.
The carcinoma pathway is via the classic adenoma-carcinoma sequence (APC/Wnt pathway), with:
- Tubular or villous adenoma architecture
- Typical (not mucinous) adenocarcinoma histology - in contrast to HNPCC/Lynch syndrome
Prophylactic colectomy is the standard of care and prevents colorectal cancer, but patients remain at risk for extracolonic neoplasia.
Upper GI and Small Bowel Pathology
| Site | Findings |
|---|---|
| Stomach | Fundic gland polyps (most common); dysplastic change occasionally; rarely adenomas |
| Duodenum | Adenomas in 60-90% of FAP patients; ampulla of Vater involved in 50-85% |
| Periampullary | 4-12% lifetime risk of duodenal/ampullary adenocarcinoma - leading cause of cancer death after prophylactic colectomy |
| Jejunum | Adenomas in ~40% |
| Ileum | Adenomas in ~20% |
The Spigelman Classification (stages I-IV) stratifies duodenal polyposis severity by polyp number, size, histology, and degree of dysplasia. Stage III/IV carries highest progression risk.
FAP Variants
Gardner Syndrome
APC germline mutations (specific mutations in codon 1310-2843 region) cause FAP plus:
- Osteomas of the mandible, skull, and long bones
- Epidermoid/sebaceous cysts
- Desmoid tumors (intra-abdominal fibromatosis) - a leading cause of morbidity
- Thyroid tumors (papillary thyroid carcinoma)
- Dental abnormalities (unerupted and supernumerary teeth)
- Congenital hypertrophy of the retinal pigment epithelium (CHRPE) - present from birth, useful as a screening marker
Turcot Syndrome
- FAP + CNS tumors
- Two-thirds have APC mutations - develop medulloblastomas
- One-third have DNA mismatch repair mutations - develop glioblastomas
Attenuated FAP (AFAP)
- Caused by mutations in the 5' and 3' regions of APC
- Fewer than 100 adenomas (typically 10-99), predominantly in the proximal colon
- Later age of polyp onset and cancer development
- Duodenal/periampullary adenomas still occur; gastric fundic gland polyps present; mandibular osteomas rare
Summary table of adenomatous polyposis syndromes
| Syndrome | Gene | Inheritance | Polyp Count | Key Extraintestinal Features |
|---|---|---|---|---|
| Classic FAP | APC | Autosomal dominant | Thousands | CHRPE, osteomas, desmoids, epidermoid cysts, thyroid tumors, medulloblastoma |
| Attenuated FAP | APC (5'/3' ends) | Autosomal dominant | <100, proximal | Rare mandibular osteomas |
| MUTYH-associated polyposis (MAP) | MUTYH (MYH) | Autosomal recessive | 5-100s, later onset | CHRPE, osteomas; sessile serrated polyps, KRAS mutations |
| Turcot syndrome (APC type) | APC | Autosomal dominant | Hundreds | Medulloblastoma |
| Turcot syndrome (MMR type) | MSH2/MLH1 | Autosomal dominant | Few | Glioblastoma |
| Polymerase proofreading-associated polyposis (PPAP) | POLD1, POLE | Autosomal dominant | Adenomas + duodenal | Endometrial cancer |
- Sleisenger and Fordtran's Gastrointestinal and Liver Disease, Table 126.9
Desmoid Tumors in FAP
Desmoid tumors (aggressive fibromatosis) occur in ~10-20% of FAP patients. They are:
- Locally invasive, non-metastasizing fibrous tumors arising from musculoaponeurotic structures
- Predominantly intra-abdominal (mesenteric) or abdominal wall in FAP
- A major cause of morbidity and post-colectomy mortality due to bowel obstruction and vascular encasement
- Strongly associated with APC mutations downstream of codon 1310
- Can be triggered by surgical trauma (post-colectomy)
Comparison with HNPCC/Lynch Syndrome (Pathology Key Points)
| Feature | FAP | HNPCC (Lynch) |
|---|---|---|
| Polyp number | Hundreds to thousands | Few (1-3) |
| Molecular pathway | APC/Wnt (CIN) | DNA mismatch repair (MSI) |
| Polyp histology | Tubular/villous adenoma | Sessile serrated adenoma |
| Carcinoma histology | Typical adenocarcinoma | Mucinous adenocarcinoma (often) |
| Carcinoma site | No predilection | Right colon predominant |
| Key gene | APC | MSH2, MLH1 |
Key Points Summary
- FAP = APC tumor suppressor gene mutation, autosomal dominant, 5q21-q22
- >100 adenomas required for classic diagnosis; can number in thousands
- Polyps histologically identical to sporadic adenomas (tubular/villous architecture)
- 100% lifetime risk of colorectal carcinoma if untreated
- Mechanism: loss of APC → beta-catenin accumulation → Wnt pathway activation → adenoma formation
- Prophylactic colectomy is standard care
- Leading extracolonic cancer risk post-colectomy: periampullary/duodenal adenocarcinoma
- Variants: Gardner syndrome (osteomas + desmoids), Turcot syndrome (medulloblastoma), AFAP (<100 polyps, proximal colon)
- MUTYH-associated polyposis (MAP) is the autosomal recessive phenocopy
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (9780443264528) | Robbins & Kumar Basic Pathology (9780323790185) | Sleisenger and Fordtran's Gastrointestinal and Liver Disease (9780323609623)
This is a shared conversation. Sign in to Orris to start your own chat.