Respiratory system pathology Chronic diffuse interstitial disease Idiopathic pulmonary fibrosis etc
Reading File
Finding Sources
Finding Sources
Searching PubMed
"idiopathic pulmonary fibrosis"[MeSH Terms] AND treatment
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Loading Image
Reading File
Reading File
Finding Sources
Loading Image
I now have all the material needed for a comprehensive, textbook-level answer. Let me compile it:
Chronic Diffuse Interstitial (Restrictive) Lung Diseases
Overview and Classification
Chronic interstitial pulmonary diseases are a heterogeneous group of disorders characterized predominantly by inflammation and fibrosis of the lung interstitium, with pulmonary function studies showing a restrictive pattern. They produce dyspnea, tachypnea, end-inspiratory crackles, and eventual cyanosis - without wheezing or airway obstruction. Typical PFT findings: reduced diffusion capacity (DLCO), reduced lung volumes, and reduced compliance. Chest radiographs show bilateral small nodules, irregular lines, or ground-glass shadows (interstitial fibrosis).
Advanced forms of all categories converge into diffuse scarring - "end-stage lung" or "honeycomb lung" - with secondary pulmonary hypertension and cor pulmonale. Most diagnoses require multidisciplinary input from clinicians, radiologists, and pathologists.
(Robbins, Cotran & Kumar - Pathologic Basis of Disease)
Major Categories (Table 15.5 - Robbins)
| Category | Diseases |
|---|---|
| Fibrosing | IPF (UIP), NSIP, Cryptogenic organizing pneumonia, CTD-associated, Pneumoconioses, Drug reactions, Radiation pneumonitis |
| Granulomatous | Sarcoidosis, Hypersensitivity pneumonitis |
| Eosinophilic | Eosinophilic pneumonia |
| Smoking-related | DIP, RB-ILD |
| Other | Langerhans cell histiocytosis, Pulmonary alveolar proteinosis, LIP |
Idiopathic Pulmonary Fibrosis (IPF) / Usual Interstitial Pneumonia (UIP)
Definition
IPF is a progressive interstitial pulmonary fibrosis of unknown etiology. Its radiologic and histologic pattern is called usual interstitial pneumonia (UIP), which is required for diagnosis. Because the cause is unknown, it is also called cryptogenic fibrosing alveolitis. The same UIP pattern may be seen secondary to connective tissue diseases, chronic hypersensitivity pneumonia, asbestosis, and drugs - making IPF a diagnosis of exclusion.
(Robbins & Kumar Basic Pathology; Grainger & Allison's Diagnostic Radiology)
Epidemiology
- Prevalence: 50-200 per 100,000
- Most common ILD of unknown cause
- Virtually never before age 50; peak in the 5th-6th decade
- Males > females
- Frequently associated with history of smoking or other environmental exposures
- Poor prognosis: estimated 50% 3- to 5-year survival
(Harrison's Principles of Internal Medicine 22E)
Pathogenesis
IPF is believed to result from repeated alveolar epithelial injury and defective repair in a genetically predisposed individual:
-
Genetic factors:
- Germline mutations in telomerase genes → cellular senescence → profibrotic phenotype
- ~35% of patients carry a variant in MUC5B (mucin production) - expressed only in lung epithelium
- Smaller number have mutations in surfactant genes (SP-C, SP-A2)
- These epithelial gene associations suggest epithelial cell abnormalities are key initiators
-
Pathologic mechanism: Abnormal epithelial repair at sites of chronic injury → exuberant fibroblastic/myofibroblastic proliferation → collagen deposition
-
TGF-β: Excessive activation drives fibrosis
-
M2 macrophages: May drive fibrosis by secreting cytokines that activate fibroblasts (IL-4, IL-13 pathway)
-
Proposed triggers: Chronic gastroesophageal reflux, environmental exposures (only small fractions of exposed individuals develop IPF, suggesting other unknown factors)
(Robbins & Kumar Basic Pathology)
Morphology
Gross:
- Pleural surfaces show cobblestoning (retraction of scars along interlobular septa)
- Cut surface: firm, rubbery, white areas of fibrosis
- Lower lobe and subpleural predominance
Histology (UIP pattern):
- Patchy interstitial fibrosis with temporal and spatial heterogeneity - the hallmark of UIP (early + late lesions coexist)
- Fibroblastic foci - earliest lesions; subepithelial collections of myofibroblasts and collagen (Fig. A above)
- Over time: dense collagenous, less cellular zones
- Honeycomb fibrosis - dense fibrosis causes alveolar wall collapse → cystic spaces lined by hyperplastic type II pneumocytes or bronchiolar epithelium
- Interstitial inflammation: lymphocytes, occasional plasma cells, mast cells, eosinophils in alveolar septae
- Secondary pulmonary hypertensive changes: intimal fibrosis, medial thickening of pulmonary arteries
- NSIP, granulomas, and hyaline membranes are absent
(Robbins, Cotran & Kumar Pathologic Basis of Disease)
The temporal heterogeneity (areas of varying fibrotic maturity) is what distinguishes UIP from NSIP, which is temporally uniform.
HRCT Findings
Classic UIP pattern on CT:
- Posterior, basilar, subpleural predominance of reticular markings
- Honeycombing (cystic spaces with thick walls)
- Traction bronchiectasis
- Minimal or absent ground-glass opacities
Findings that should suggest an alternative diagnosis: extensive ground-glass opacities, bronchovascular distribution, micronodules, mosaic attenuation, upper lung predominance, or subpleural sparing.
(Harrison's 22E)
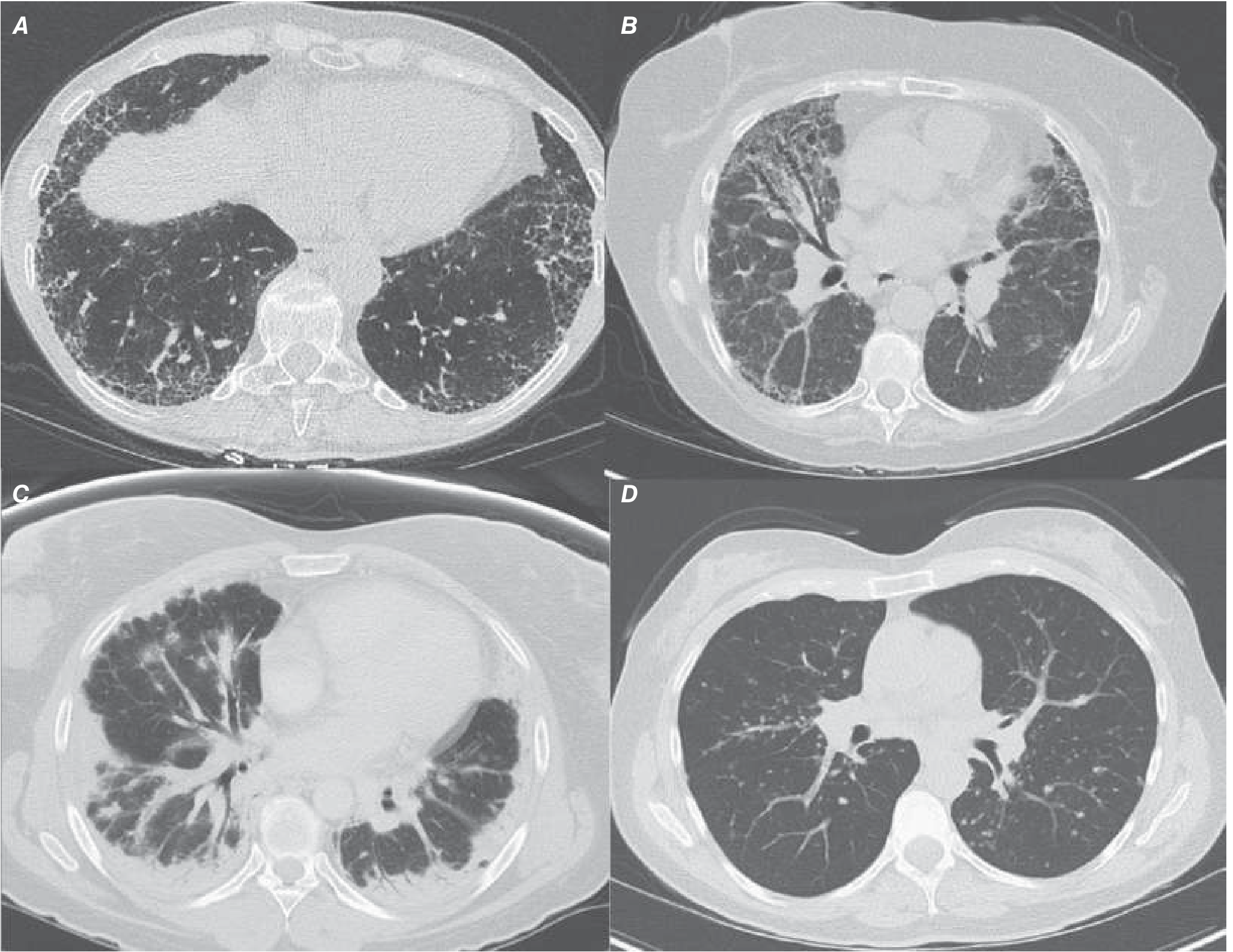
Clinical Features
- Gradual onset of nonproductive cough and progressive dyspnea
- "Velcro-like" crackles (dry, end-inspiratory) on auscultation - characteristic
- Late stages: cyanosis, cor pulmonale, peripheral edema
- Cough is an independent predictor of progression; refractory to standard antitussives
- PFTs: restrictive pattern (↓ TLC, ↓ FVC, preserved FEV1/FVC, ↓ DLCO)
Diagnosis
- Clinical features + HRCT showing UIP pattern is often sufficient
- Surgical VATS lung biopsy when needed: shows subpleural reticulation, honeycomb changes, fibroblastic foci, temporal heterogeneity (UIP pattern)
- Multidisciplinary discussion (MDT) of clinicians, radiologists, and pathologists is standard practice
Treatment
Historically felt to be refractory to medical therapy; lung transplantation was the only viable option. This changed in 2014 with large RCTs:
- Antifibrotic therapy (pirfenidone and nintedanib) - slows decline in lung function; meta-analyses suggest may also improve survival
- A 2024 meta-analysis (PMID 38963453) confirmed real-world safety and effectiveness of pirfenidone and nintedanib
- Recent trials suggest antifibrotics may benefit other forms of progressive pulmonary fibrosis beyond IPF
- Immunosuppression (previously prescribed widely) has been shown to be associated with increased morbidity and mortality - no longer recommended
- Physical therapy + supplemental oxygen: improve exercise tolerance, reduce pulmonary hypertension risk
- Lung transplantation: extends survival in selected patients
Other Key Idiopathic Interstitial Pneumonias (IIPs)
Nonspecific Interstitial Pneumonia (NSIP)
| Feature | Detail |
|---|---|
| Demographics | Nonsmoking females, 5th decade; frequently CTD-associated |
| HRCT | Diffuse bilateral symmetric ground-glass + reticular opacities, lower zone; subpleural sparing; no honeycombing |
| Histology | Uniform inflammation and fibrosis (temporally uniform); no fibroblastic foci or honeycombing |
| Prognosis | Better than IPF; 5-year survival >80% (cellular pattern > fibrotic) |
| Treatment | Steroids, cytotoxics (mycophenolate, azathioprine, cyclophosphamide), biologics (rituximab, tocilizumab) |
Key distinction from IPF: temporal uniformity - NSIP lacks the spatial heterogeneity of UIP.
(Harrison's 22E; Murray & Nadel's)
Cryptogenic Organizing Pneumonia (COP)
- Formerly called BOOP (bronchiolitis obliterans organizing pneumonia)
- Patients in 50s-60s; subacute flu-like illness with cough, dyspnea, fever, fatigue
- HRCT: Patchy, sometimes migratory subpleural consolidative opacities ± ground-glass; "reversed halo sign" (atoll sign)
- Histology: Plugs of organizing fibrous tissue (Masson bodies) within alveolar ducts and alveoli
- Can be secondary to CTD, medications, or malignancy
- Generally responsive to corticosteroids
Respiratory Bronchiolitis-ILD (RB-ILD) and Desquamative Interstitial Pneumonia (DIP)
- Smoking-related ILDs - occur in active/heavy smokers, typically ages 40-50
- RB-ILD histology: Pigmented macrophages in respiratory bronchioles and alveolar ducts, peribronchiolar fibrosis; patchy and peribroncheolar
- DIP histology: Diffuse pigmented macrophage accumulation throughout alveoli, pneumocyte hyperplasia, prominent interstitial thickening
- Key treatment: Smoking cessation (most important intervention)
Granulomatous ILD
Sarcoidosis (in the context of ILD)
- Noncaseating granulomas in lung interstitium and lymph nodes
- HRCT: Hilar + mediastinal lymphadenopathy; bronchovascular nodular pattern; miliary nodules
- Serum ACE elevated; BAL shows lymphocytosis with CD4:CD8 ratio >3.5
- Variable - may remit spontaneously or progress to fibrosis
Hypersensitivity Pneumonitis (Extrinsic Allergic Alveolitis)
- Immune-mediated (Type III + Type IV hypersensitivity) response to inhaled organic antigens
- Examples: Farmer's lung (thermophilic actinomycetes), Bird fancier's lung
- Histology: poorly-formed granulomas, organizing pneumonia, lymphocytic alveolitis
- UIP-like pattern can develop in chronic HP
ILD Classification by Histologic Pattern (Summary Table)
(Grainger & Allison's Diagnostic Radiology, Table 9-2)
| IIP Entity | Histological Pattern | Key HRCT Features |
|---|---|---|
| IPF | UIP | Subpleural reticular + honeycombing + traction bronchiectasis |
| NSIP | NSIP | Bilateral symmetric ground-glass + reticular; subpleural sparing |
| COP | Organizing pneumonia | Patchy subpleural consolidation; reversed halo sign |
| AIP (Hamman-Rich) | Diffuse alveolar damage | Ground-glass ± traction bronchiectasis |
| RB-ILD | Respiratory bronchiolitis | Centrilobular nodules + ground-glass; no honeycombing |
| DIP | DIP | Diffuse bilateral ground-glass |
| LIP | LIP | Ground-glass + thin-walled cysts |
Histopathology of ILD - Key Images
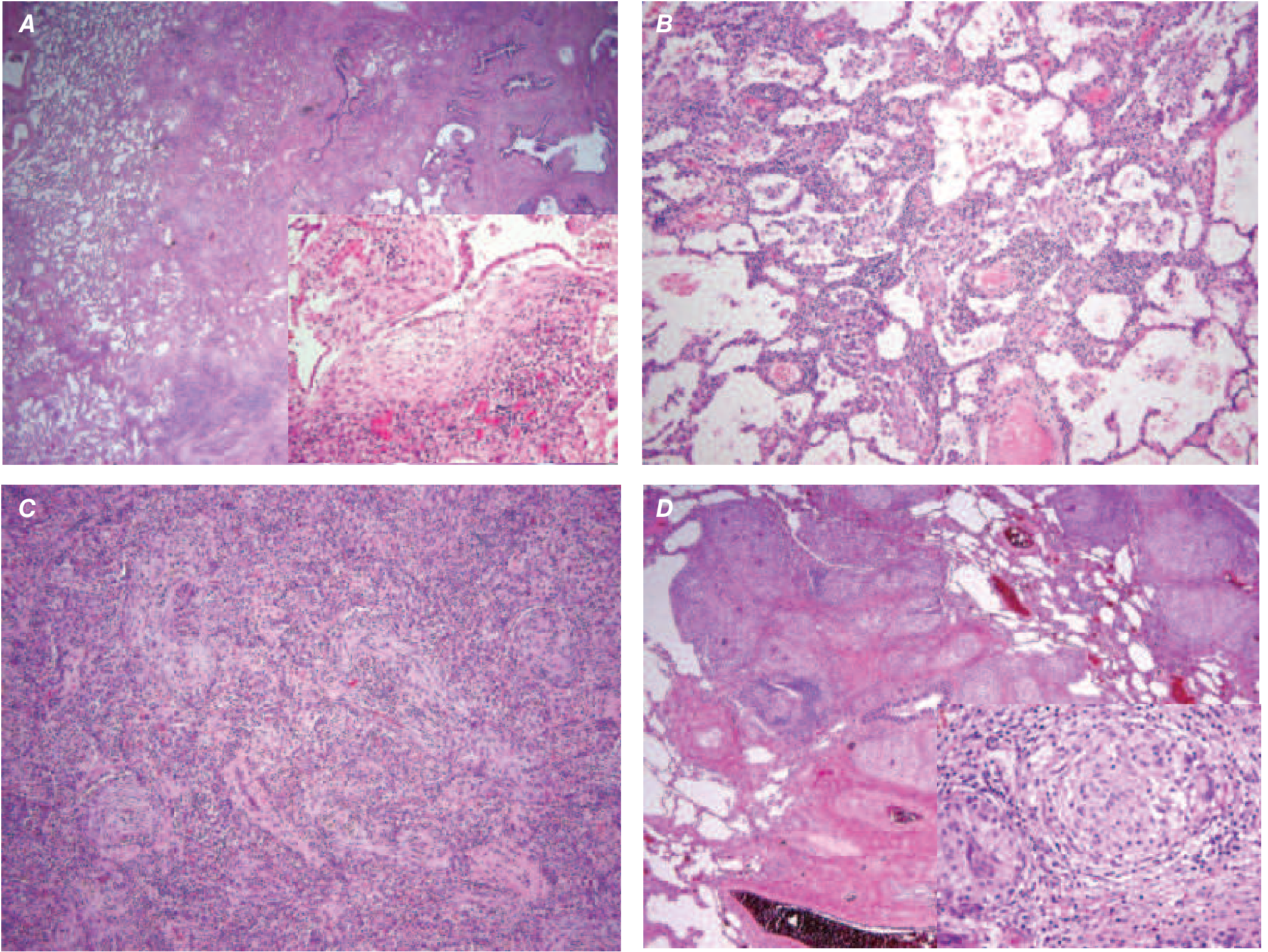
Key Pathologic Comparisons: IPF vs NSIP
| Feature | IPF (UIP) | NSIP |
|---|---|---|
| Temporal pattern | Heterogeneous (old + new lesions coexist) | Uniform (all same age) |
| Fibroblastic foci | Prominent | Absent or rare |
| Honeycombing | Present | Absent or scanty |
| Inflammation | Scant | More prominent (cellular form) |
| Subpleural sparing | Absent | Often present |
| Prognosis | Poor (50% 5-yr survival) | Better (>80% 5-yr survival) |
Recent Evidence (PubMed, 2024-2025)
- A 2024 systematic review/meta-analysis (PMID 38963453) confirmed real-world safety and effectiveness of pirfenidone and nintedanib in IPF treatment
- A 2025 meta-analysis (PMID 40107663) examined comorbidities in IPF and progressive pulmonary fibrosis trial populations, noting their significant prevalence and impact
- A 2025 transcriptomics meta-analysis (PMID 39603671) of ILD identified molecular signatures distinguishing ILD subtypes, which may inform future precision therapy
Sources: Robbins, Cotran & Kumar Pathologic Basis of Disease (2024) | Robbins & Kumar Basic Pathology | Harrison's Principles of Internal Medicine 22E (2025) | Murray & Nadel's Textbook of Respiratory Medicine | Grainger & Allison's Diagnostic Radiology
This is a shared conversation. Sign in to Orris to start your own chat.