IPF with excerbation full details for md exams 15marks answer
Reading File
Finding Sources
Finding Sources
Searching PubMed
"idiopathic pulmonary fibrosis" AND "acute exacerbation"
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Finding Sources
Loading Image
Loading Image
I now have comprehensive, authoritative content from multiple major textbooks. Let me compile the full MD exam answer.
Idiopathic Pulmonary Fibrosis (IPF) with Acute Exacerbation - Full Details for MD Examination
1. DEFINITION
Idiopathic Pulmonary Fibrosis (IPF) is a specific form of chronic, progressive, fibrosing interstitial pneumonia of unknown cause, limited to the lungs, occurring primarily in older adults, and associated with the histopathological and/or radiological pattern of usual interstitial pneumonia (UIP). It is progressive, irreversible, and invariably fatal.
Acute Exacerbation of IPF (AE-IPF) is defined (per the International Working Group Report) as: "an acute clinically significant respiratory deterioration characterized by evidence of new widespread alveolar abnormality" occurring in a patient with known IPF, typically within less than 1 month of onset, without a fully explanatory alternative cause such as cardiac failure or fluid overload.
2. EPIDEMIOLOGY
| Parameter | Data |
|---|---|
| Incidence (Europe/North America) | 3-9 cases/100,000/year |
| US prevalence | 0.8-65/100,000 |
| Peak age | >60 years (most commonly 5th-6th decade) |
| Sex | Male > Female |
| AE-IPF annual incidence | 4-15% of IPF patients per year |
| Median survival (IPF) | 3-5 years from diagnosis (~50% 3-5 year survival) |
- Incidence and prevalence increase markedly with age, particularly after 75 years
- Worldwide IPF mortality has increased steadily
- Murray & Nadel's Textbook of Respiratory Medicine; Harrison's Principles of Internal Medicine 22E
3. ETIOPATHOGENESIS
Risk Factors
- Cigarette smoking - strongest environmental risk factor
- Age >60 years
- Male sex
- Gastroesophageal reflux disease (GERD) - microaspiration hypothesis
- Viral infections (herpesvirus, EBV, CMV)
- Occupational dust exposure (metal, wood dust)
Genetic / Familial IPF
- Familial pulmonary fibrosis (FPF): identified in ~15% as familial clusters
- Gene mutations: SFTPC (surfactant protein C), SFTPA2 (surfactant protein A2), MUC5B (mucin 5B)
- ~15% of FPF cases: short telomere syndrome from mutations in TERC, TERT (telomerase components) - may present with bone marrow failure, premature graying, cirrhosis, nail dystrophy, mucosal leukoplakia
Pathogenesis (Current Model)
The pathophysiology centers on repeated alveolar epithelial injury with dysregulated repair, not primary inflammation:
- Epithelial injury - repeated microinjuries to alveolar epithelial cells (type II pneumocytes)
- Abnormal wound healing - failure of normal regeneration
- Fibroblast activation - myofibroblast proliferation and collagen deposition
- Fibroblast foci formation - subepithelial collections of myofibroblasts; hallmark of active fibrogenesis
- Progressive architectural destruction - honeycombing, traction bronchiectasis
Molecular mediators: TGF-β (central fibrogenic cytokine), PDGF, VEGF, matrix metalloproteinases, aberrant Wnt signaling.
- Murray & Nadel; Washington Manual of Medical Therapeutics
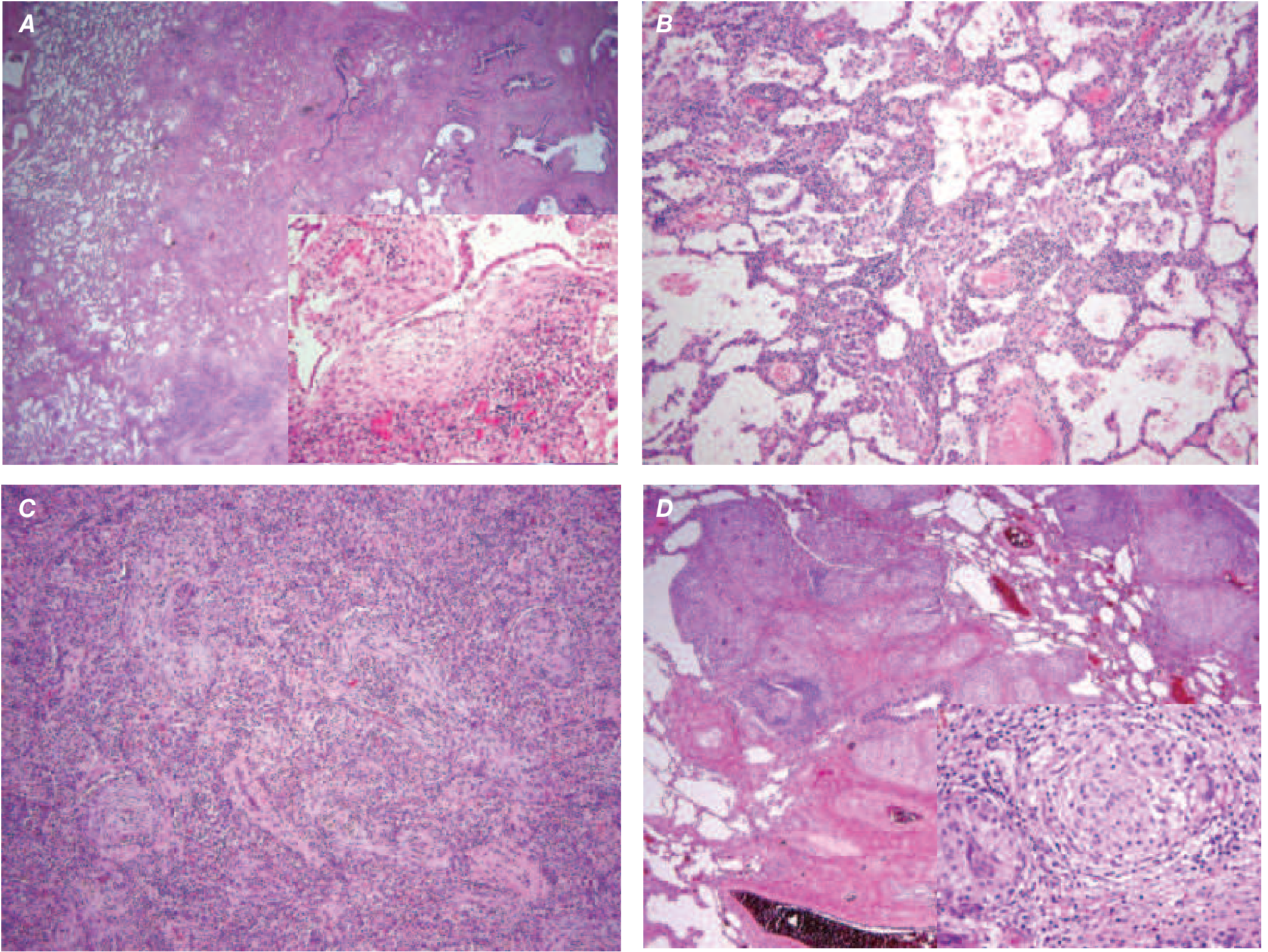
4. HISTOPATHOLOGY - UIP PATTERN
The histopathologic pattern is Usual Interstitial Pneumonia (UIP), characterized by:
- Temporal and spatial heterogeneity - areas of dense fibrosis alternating with normal lung
- Subpleural and paraseptal distribution - lower lobe predominance
- Fibroblast foci - discrete subepithelial collections of myofibroblasts and collagen (marker of active fibrogenesis)
- Honeycombing - enlarged cystic airspaces lined by bronchiolar epithelium, often mucin-filled
- Minimal inflammation
- Gross appearance: nodular pleural surface resembling hepatic cirrhosis ("cobblestone" or "shaggy" appearance)
5. CLINICAL FEATURES
Symptoms
- Slowly progressive dyspnea - over months to years (insidious onset)
- Non-productive cough (dry, persistent)
- Rare systemic or extrapulmonary symptoms
- Constitutional symptoms (fatigue, weight loss) may occur with advanced disease
Signs
- Bibasilar fine, dry (Velcro) inspiratory crackles - most characteristic
- Digital clubbing - 45-75% of patients
- Features of cor pulmonale in advanced disease (elevated JVP, right heart failure, peripheral edema)
- Cyanosis in late disease
6. INVESTIGATIONS
Pulmonary Function Tests (PFTs)
- Restrictive ventilatory defect: reduced TLC, FVC, FEV1 (with preserved or elevated FEV1/FVC ratio)
- Reduced DLCO (diffusing capacity for CO) - often disproportionately low; correlates with prognosis
- Progressive FVC decline >10% per year = marker of disease progression and poor prognosis
6-Minute Walk Test (6MWT)
- Reduced exercise tolerance
- Desaturation during exercise (SpO2 <88%) is common
- Used for transplant listing criteria
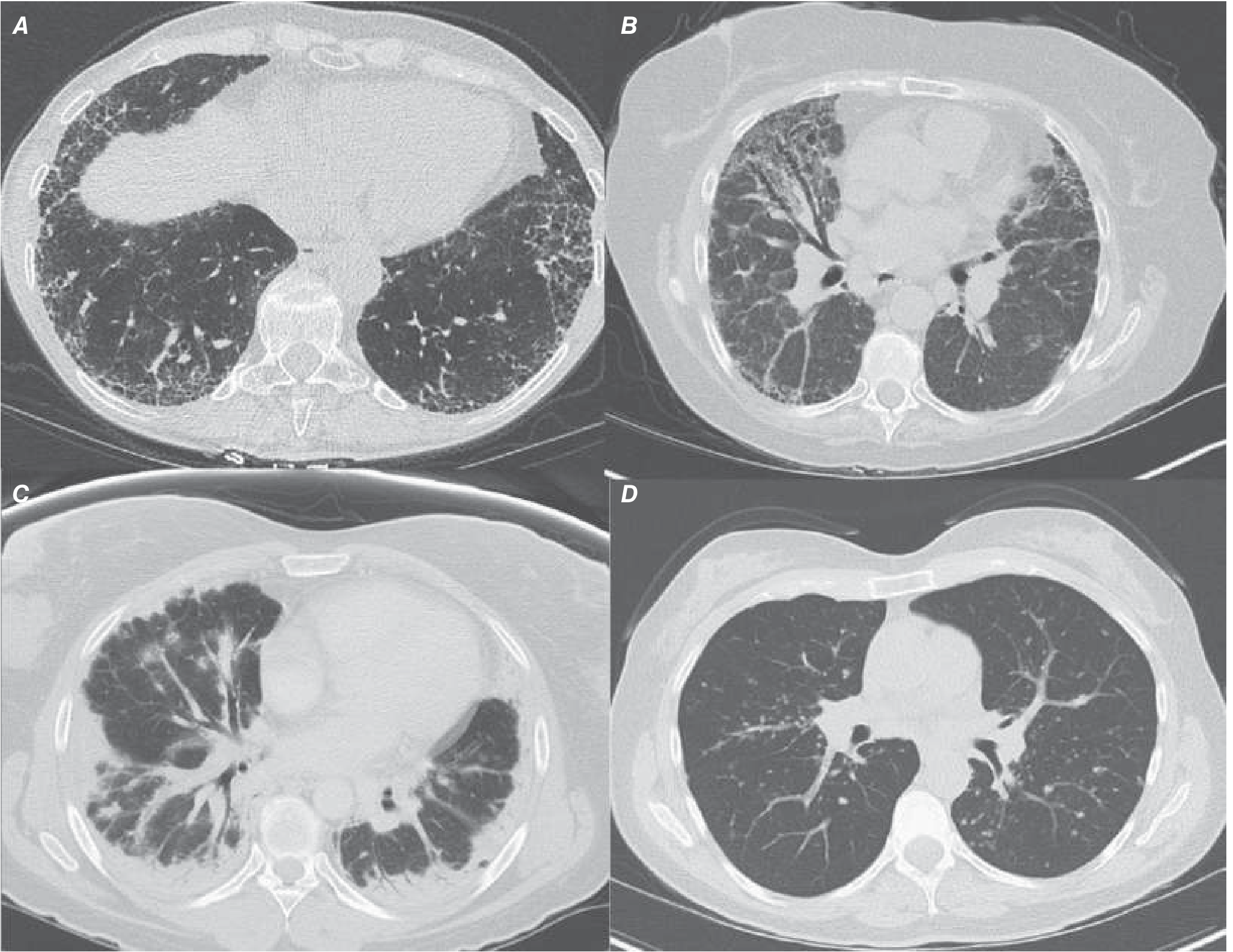
Chest HRCT (High-Resolution CT) - MANDATORY
The definite UIP pattern on HRCT includes:
- Bilateral, symmetric, subpleural, basilar-predominant reticulation
- Honeycombing ± traction bronchiectasis
- Geographic or heterogeneous involvement
- No features suggesting an alternative diagnosis
Atypical features (suggest alternative diagnosis):
- Extensive ground-glass opacities
- Upper or mid-lung predominance
- Micronodules
- Bronchovascular distribution
- Mosaic attenuation
Blood/Serological Tests
- ANA, rheumatoid factor, anti-CCP: to exclude connective tissue disease (CTD)
- Hypersensitivity precipitins: to exclude hypersensitivity pneumonitis (HP)
- CBC, metabolic panel: baseline and comorbidity assessment
- ABG: hypoxemia (type 1 respiratory failure) at rest or exercise
Bronchoalveolar Lavage (BAL)
- Used to exclude other diagnoses (infection, malignancy, eosinophilia)
- BAL cell differential in IPF: may show slightly elevated neutrophils/eosinophils
- Not diagnostic for IPF
Lung Biopsy
- VATS (Video-Assisted Thoracoscopic Surgery) lung biopsy is preferred if biopsy is needed
- Transbronchial biopsy (TBBx) is diagnostic in <1/3 of cases - not preferred
- Cryobiopsy is an emerging alternative
- Biopsy needed if HRCT pattern is not "definite UIP" in appropriate clinical context
7. DIAGNOSIS OF IPF
Diagnosis requires:
- Exclusion of all other causes of fibrosing lung disease - CTD, hypersensitivity pneumonitis, drug toxicity, sarcoidosis
- Either:
- A definite UIP pattern on HRCT (sufficient without biopsy in the right clinical context), OR
- A UIP pattern on surgical lung biopsy (when HRCT is indeterminate)
- Multidisciplinary discussion (MDD) among pulmonology, radiology, and pathology specialists with ILD expertise increases diagnostic accuracy
- Washington Manual of Medical Therapeutics; Harrison's 22E
8. TREATMENT OF IPF (Stable Disease)
Disease-Modifying (Antifibrotic) Therapy
| Drug | Mechanism | Evidence |
|---|---|---|
| Pirfenidone | Inhibits TGF-β, PDGF, FGF; anti-inflammatory, antifibrotic | Slows FVC decline; CAPACITY, ASCEND trials |
| Nintedanib | Triple tyrosine kinase inhibitor (VEGFR, PDGFR, FGFR) | Slows FVC decline; INPULSIS trials (2014) |
Both agents are first-line; no head-to-head superiority. Meta-analyses suggest both may also improve survival.
What NOT to Use
- Prednisone + azathioprine + NAC: PANTHER-IPF trial showed increased mortality and hospitalization - now contraindicated
- Interferon-γ: INSPIRE trial - no survival benefit
- Warfarin: no benefit, potential harm
Supportive / Adjunctive Care
- Supplemental oxygen (when SpO2 <88% at rest or exercise) - reduces pulmonary hypertension risk
- Pulmonary rehabilitation - improves exercise tolerance
- PPI / antacid therapy for GERD (weak recommendation; data mixed)
- Pneumococcal and influenza vaccination
- Treatment of comorbidities (pulmonary hypertension, OSA, lung cancer, DVT/PE)
- Palliative care for dyspnea (opioids, benzodiazepines)
Lung Transplantation
- Indicated for: significant FVC decline, oxygen dependence, FVC <80% predicted, DLCO <40% predicted
- ISHLT criteria for referral; bilateral lung transplant preferred
- Extends survival and improves quality of life in selected patients
9. ACUTE EXACERBATION OF IPF (AE-IPF)
Definition (International Working Group Report, 2016)
"An acute clinically significant respiratory deterioration characterized by evidence of new widespread alveolar abnormality"
Diagnostic Criteria (All 4 must be met)
- Previous or concurrent diagnosis of IPF
- Acute worsening or development of dyspnea - typically <1 month duration
- CT findings: new bilateral ground-glass opacity and/or consolidation superimposed on a background UIP pattern
- Deterioration not fully explained by cardiac failure or fluid overload
Classification
- AE-IPF with identifiable trigger: respiratory infection (bacterial/viral), aspiration, drug toxicity, surgical procedure (esp. post-lung resection)
- AE-IPF without identifiable trigger (idiopathic AE-IPF): many acute episodes have no identified cause
(Note: The updated 2016 definition includes episodes WITH an identifiable trigger, unlike the earlier definition which required exclusion of all triggers)
Epidemiology
- Annual incidence: 4-15% of IPF patients
- Mortality: >50% in-hospital mortality; 1-year mortality approaches 90% in some series
- Some deaths in IPF are directly attributed to acute exacerbation episodes
Pathogenesis of AE-IPF
- Exact mechanism unclear
- Proposed triggers/mechanisms:
- Occult viral infection (herpesvirus family)
- Microaspiration
- Disordered epithelial cell integrity
- Acute inflammation with excessive cytokines and matrix metalloproteinases (MMPs)
- Antifibrinolytic alveolar environment
- Surgical stress or anesthesia
Histopathology of AE-IPF
- Diffuse alveolar damage (DAD) superimposed on background UIP - most common pattern
- Occasionally: organizing pneumonia pattern, extensive fibroblastic foci
- Histologic evaluation is not required to diagnose AE-IPF, but biopsy/autopsy will show the above
Clinical Features
- Rapid (<1 month) worsening of dyspnea
- Worsening hypoxemia (progressive desaturation, increasing oxygen requirements)
- New bilateral crackles on new areas of the lung
- Fever may or may not be present (fever more suggestive of infection)
- Tachycardia, tachypnea
HRCT Findings in AE-IPF
- New bilateral ground-glass opacities and/or consolidation
- Superimposed on the background UIP pattern (honeycombing + subpleural reticulation)
- Diffuse (more than 2 lobes), or multifocal distribution
- Must be distinguished from: pulmonary edema, atypical infection, diffuse alveolar hemorrhage
Management of AE-IPF
Step 1 - Exclude and treat treatable causes:
- Broad-spectrum antibiotics (cover atypicals) if infection cannot be excluded
- Rule out pulmonary embolism (CT-PA if clinically suspected)
- Rule out heart failure (echo, BNP)
- BAL if feasible - to exclude infection
Step 2 - Supportive Care:
- Supplemental oxygen
- Mechanical ventilation - controversial; poor outcomes; not recommended unless as bridge to lung transplantation
- High-flow nasal cannula (HFNC) or non-invasive ventilation (NIV) may be tried
- Thromboprophylaxis
Step 3 - Pharmacological Treatment (after excluding infection):
| Drug | Dose/Regimen | Evidence |
|---|---|---|
| IV Methylprednisolone (pulse) | 0.5-1 g/day IV for 3 days, then taper to oral prednisone | Commonly used; no RCT; expert consensus |
| + Cyclophosphamide | May be added | Anecdotal; no proven benefit |
| + Cyclosporine | May be added | Anecdotal |
| Nintedanib | Continue/initiate | May reduce rate of future AEs (post-hoc INPULSIS data) |
- No RCT has demonstrated benefit for any treatment in AE-IPF
- Rationale for steroids: suppress cellular/humoral immunity, reduce acute inflammation
- Current guidelines: weak recommendation to use corticosteroids in AE-IPF; appropriate dose, route, and duration unknown
- Direct hemoperfusion with polymyxin B-immobilized fiber column (PMX-F): some reports of benefit (binds endotoxin, cytokines, neutrophil elastase) - not standard therapy
PANTHER-IPF warning: The combination of prednisone + azathioprine + NAC is contraindicated in stable IPF; however, high-dose pulse IV methylprednisolone is still used as empirical therapy for AE-IPF.
Prognosis of AE-IPF
- Very poor - in-hospital mortality >50%, often approaching 70-80%
- Median survival after AE-IPF: weeks to a few months
- Survivors often have accelerated progressive decline
- AE-IPF represents a major cause of IPF-related death
- Murray & Nadel's Textbook of Respiratory Medicine, pp. 2027; Fishman's Pulmonary Diseases; Harrison's 22E; Washington Manual
10. COMORBIDITIES IN IPF
| Comorbidity | Clinical significance |
|---|---|
| Pulmonary hypertension (IPF-PH) | 32-44% of IPF patients; mPAP >25 mmHg; markedly worsens prognosis (median survival <1 year if PASP >50 mmHg) |
| GERD | Associated with microaspiration; may trigger AE-IPF; weak recommendation to treat |
| Obstructive sleep apnea (OSA) | Prevalent; worsens hypoxemia |
| Lung cancer | IPF is an independent risk factor for lung cancer (5-10x increased risk) |
| Combined pulmonary fibrosis and emphysema (CPFE) | PH prevalence 30-50%; markedly poor outcomes |
| Venous thromboembolism (DVT/PE) | Increased risk; often precipitates AE-IPF |
11. DIFFERENTIAL DIAGNOSIS
| Disease | Key distinguishing features |
|---|---|
| NSIP (Non-specific interstitial pneumonia) | Subpleural sparing, symmetric GGO, younger females, CTD association |
| COP (Cryptogenic organizing pneumonia) | Patchy consolidative opacities, responsive to steroids |
| Hypersensitivity pneumonitis (HP) | Upper/mid-lung, exposure history, positive precipitins |
| Sarcoidosis | Upper lobe, lymphadenopathy, non-caseating granulomas |
| CTD-associated ILD | Systemic features, serological markers |
| Drug-induced ILD | Drug history (amiodarone, methotrexate, etc.) |
12. PROGNOSIS
- IPF carries a poor prognosis: median survival 3-5 years from diagnosis
- 50% 3-5 year survival rate
- Disease course: slow progressive decline in most; some have accelerating decline punctuated by AE episodes
- Poor prognostic factors:
- FVC decline >10% in 6 months
- DLCO <40% predicted
- SpO2 <88% on 6MWT
- Pulmonary hypertension
- Honeycombing on HRCT
- AE-IPF episodes
- GAP score (Gender, Age, Physiology) - validated prognostic model
13. SUMMARY TABLE: AE-IPF AT A GLANCE
| Feature | Detail |
|---|---|
| Definition | Acute respiratory deterioration (<1 month) with new bilateral alveolar opacities superimposed on UIP |
| Incidence | 4-15%/year |
| Triggers | Infection, aspiration, drugs, surgery, idiopathic |
| Histology | DAD on UIP background |
| HRCT | New bilateral GGO/consolidation on background UIP |
| Treatment | IV methylprednisolone ± immunosuppressants (no RCT data); supportive care |
| Mechanical ventilation | Controversial; avoid unless bridge to transplant |
| Prognosis | In-hospital mortality >50%; catastrophic outcome |
Recent Evidence Update (2023-2025)
A 2025 systematic review (PMID: 39721758) on corticosteroid therapy for acute exacerbation of ILDs found that evidence remains observational and weak - no RCTs exist, consistent with textbook guidance. A 2024 narrative review (PMID: 39144320) highlights that despite new treatment modalities being explored, corticosteroids and antifibrotics remain the mainstay, and a 2024 meta-analysis (PMID: 38963453) confirms the real-world safety and effectiveness of pirfenidone and nintedanib.
Sources: Murray & Nadel's Textbook of Respiratory Medicine (2-Volume); Harrison's Principles of Internal Medicine 22E (2025); Washington Manual of Medical Therapeutics; Fishman's Pulmonary Diseases and Disorders; Grainger & Allison's Diagnostic Radiology
This is a shared conversation. Sign in to Orris to start your own chat.