Pathogenesis of h pylori
Reading File
Finding Sources
Finding Sources
Searching PubMed
"Helicobacter pylori"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive material from multiple authoritative sources. Let me compile a thorough, well-organized response.
Pathogenesis of Helicobacter pylori
1. The Organism
H. pylori is a slender, curved, Gram-negative rod with multiple polar flagella that confer rapid motility in viscous mucus. It grows slowly (3-5 days) in a microaerophilic atmosphere. Discovery by Warren and Marshall in 1982 fundamentally changed the understanding of peptic ulcer disease. Roughly half the global population is infected; the organism is acquired in childhood, typically via the fecal-oral route, and persists for life without treatment.
2. How H. pylori Survives in the Stomach
The gastric lumen has a pH of 1-3, which would normally kill bacteria. H. pylori overcomes this through several coordinated mechanisms:
| Mechanism | Detail |
|---|---|
| Urease | Produced in enormous quantities (~6% of total bacterial protein). Hydrolyzes urea → NH₃ + CO₂. Ammonia neutralizes local acid, creating a pH microenvironment of ~6-7 around the bacterium. Also acts as a chemoattractant for host phagocytes. |
| Flagellar motility | Allows the bacterium to move through the viscous mucus layer toward the epithelial surface, where pH is near-physiological (5.0-7.0). |
| Mucus penetration | Decreases mucin synthesis, thinning the protective mucous layer and facilitating bacterial access to the epithelium. |
3. Colonization and Adhesion
H. pylori colonizes only gastric-type epithelium (not intestinal-type). Key outer membrane proteins (adhesins) mediate tight attachment:
- BabA (blood-antigen binding protein A): binds fucosylated Lewis b antigen on gastric epithelial cells.
- SabA: binds sialyl Lewis X receptors, particularly upregulated in inflamed mucosa.
Adhesion is essential - strains lacking functional adhesins cannot efficiently colonize. H. pylori does not invade the gastric mucosa; it remains in the mucus layer overlying surface epithelial cells.
4. Virulence Factors: The Key Players
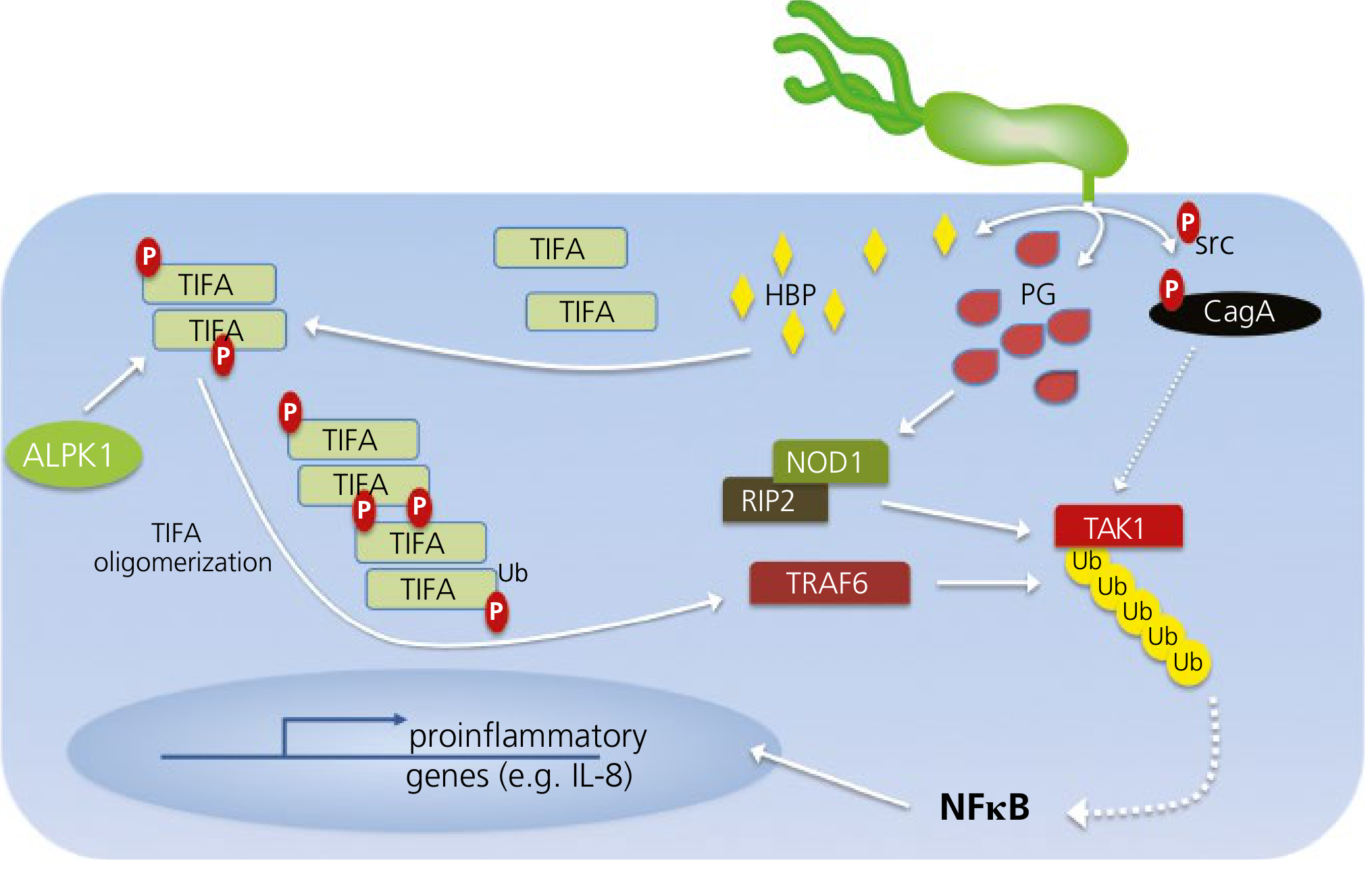
A. CagA and the Cag Pathogenicity Island (cag PAI)
This is the most important virulence determinant. The cag PAI is a ~40 kb DNA segment encoding a Type IV Secretion System (T4SS) - a molecular "injection needle" that translocates effector molecules directly into host epithelial cells.
CagA (cytotoxin-associated gene A) is the terminal effector:
- Injected into the host cell cytoplasm via the T4SS
- Tyrosine-phosphorylated by Src and Abl kinases inside the host cell
- Phospho-CagA activates the tyrosine phosphatase SHP-2, triggering morphological transformations resembling growth factor stimulation
- Non-phosphorylated CagA interacts with E-cadherin, c-Met receptor, and activates β-catenin → disrupts cell-cell junctions, causes loss of cell polarity, and drives mitogenic responses
- Increases spermine oxidase (SMO) production → oxidative DNA damage → selects for apoptosis-resistant cells → carcinogenesis
- Targets tumor suppressor p53 to dysregulate apoptosis
- Activates NF-κB → IL-8 and proinflammatory gene expression → neutrophil recruitment
The cag PAI is present in ~50% of H. pylori isolates overall, but in ~90% of strains in high-gastric-cancer-prevalence populations (e.g., East Asia). CagA-positive strains more effectively colonize the gastric body (in addition to the antrum) and induce greater inflammation, atrophy, and intestinal metaplasia.
The T4SS also translocates peptidoglycan (PG) fragments and heptose bisphosphate (HBP) into host cells:
- PG activates NOD1 → RIP2 → TRAF6 → TAK1 → NF-κB → IL-8
- HBP activates ALPK1 → TIFA oligomerization → NF-κB → IL-8
B. VacA (Vacuolating Cytotoxin A)
- Secreted protein that enters host cells and forms channels in lysosomal and endosomal membranes, generating multiple large cytoplasmic vacuoles
- Induces apoptosis in epithelial cells
- Disrupts the balance between cell death and proliferation
- Acts as an activator of IL-8-mediated acute inflammation
- VacA i1 strains (inner region allele 1) strongly associate with cagA positivity and gastric cancer risk
- Also delivered to host cells by the T4SS
C. Other Virulence Factors
| Factor | Function |
|---|---|
| Urease | Beyond acid neutralization: chemoattractant and activator of host phagocytic/inflammatory cells |
| Mucinase & phospholipase | Degrades the mucous layer, disrupts the hydrophobic phospholipid barrier |
| Neutrophil-activating protein A (NapA) | Recruits and activates neutrophils |
| Heat shock protein 60 | Contributes to inflammation |
| OipA (outer inflammatory protein) | Induces IL-8 production independently of CagA |
5. The Inflammatory Response
H. pylori infection is not invasive, yet it triggers a vigorous host immune response:
- Acute phase: Bacterial adherence triggers IL-8 release from epithelial cells → recruits neutrophils → "active gastritis"
- Chronic phase: Sequential recruitment of T and B lymphocytes, plasma cells, and macrophages
- Cytokine cascade: Enhanced expression of TNF-α, IL-1β, IL-6, IL-8, and IL-12
- Immune evasion: Despite this response, H. pylori is not cleared - it down-regulates the Th1/Th17 response and evades phagocytic killing
The result is a chronic active gastritis that persists lifelong without treatment.
6. How Gastritis Leads to Disease
The pattern of gastritis distribution determines clinical outcome - this is the key concept:
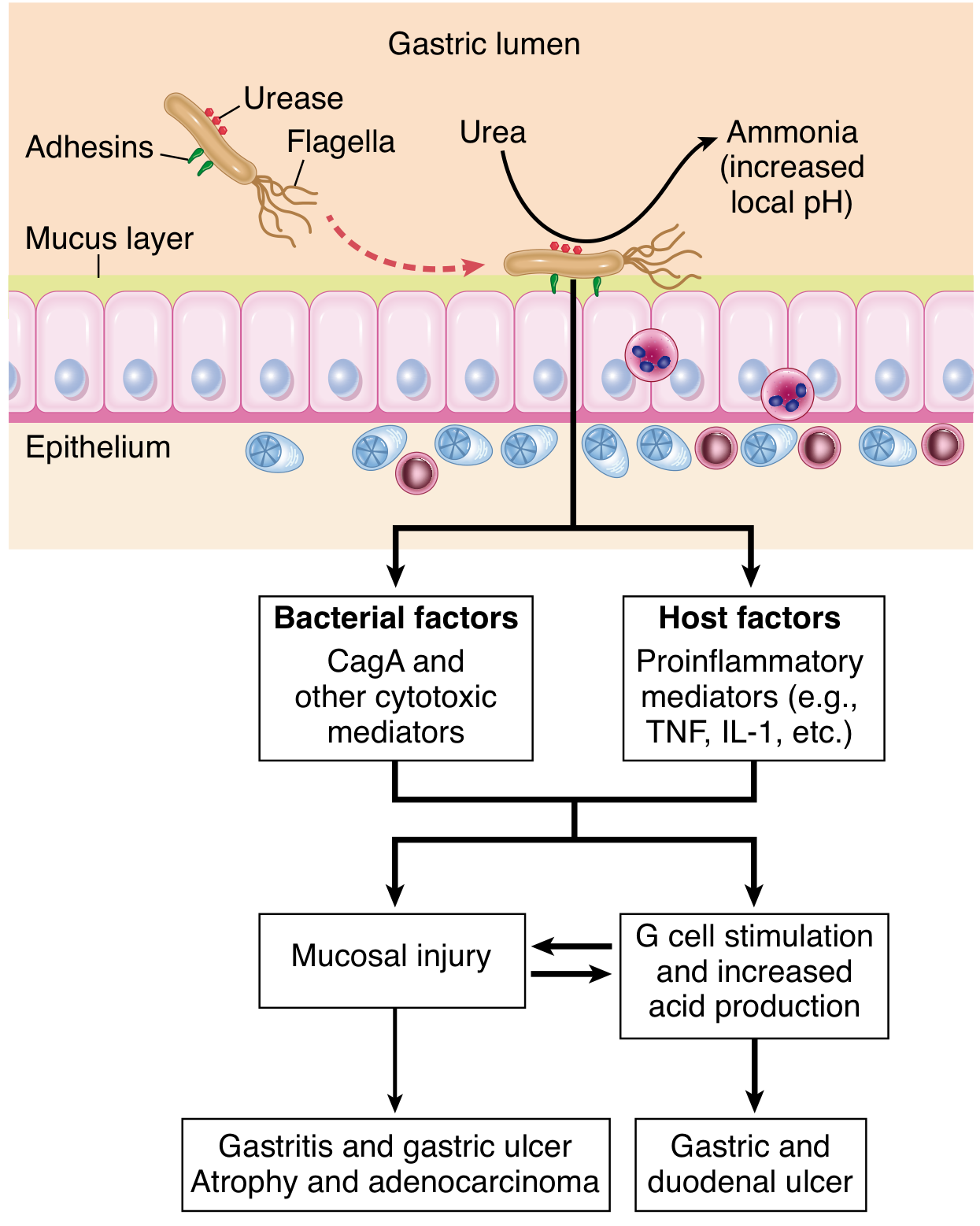
Antral-Predominant Gastritis → Peptic Ulcer Disease (PUD)
- Antral gastritis → local inflammation → inhibits D cells (which normally produce somatostatin) → loss of inhibition on G cells → hypergastrinemia
- Excess gastrin stimulates parietal cells in the corpus → increased acid secretion (hyperchlorhydria)
- High acid load damages the duodenal bulb → gastric metaplasia of duodenal epithelium → H. pylori colonizes the metaplastic patches → duodenitis → duodenal ulcer (DU)
- NSAIDs synergize by disrupting the prostaglandin-mediated mucosal defence
- Lifetime risk of PUD in infected individuals: ~10-20%
Pangastritis / Corpus-Predominant Gastritis → Atrophy → Gastric Cancer
- Long-standing infection extends to the gastric body and fundus
- Atrophic gastritis with loss of parietal cells → hypochlorhydria (paradoxically reduces ulcer risk)
- Intestinal metaplasia (mucous cells replaced by intestinal-type cells)
- Progression: chronic gastritis → atrophy → intestinal metaplasia → dysplasia → gastric adenocarcinoma (Correa cascade)
- H. pylori is classified as a Group 1 (definite) carcinogen by the IARC
- CagA-positive strains are more strongly linked to cancer
Gastric MALToma (Mucosa-Associated Lymphoid Tissue)
- Normally the stomach has no lymphoid tissue
- Chronic H. pylori infection induces polyclonal B-cell proliferation in the gastric mucosa (acquired MALT)
- Ongoing antigenic stimulation may lead to acquisition of driver mutations → monoclonal gastric B-cell MALT lymphoma
- Early-stage MALToma may regress with H. pylori eradication alone
7. Host Genetic Factors
Outcome is not determined by the bacterium alone. Host polymorphisms matter:
- IL-1β and IL-1RN polymorphisms: increased IL-1β expression → stronger acid suppression → more favorable environment for corpus colonization → greater atrophy risk
- TNF-α polymorphisms: increased TNF production → enhanced mucosal damage
- IL-10 polymorphisms: decreased IL-10 (anti-inflammatory) → more aggressive inflammation
- These polymorphisms help explain why only ~10-20% of infected individuals develop PUD and <1% develop gastric cancer
8. Summary Flowchart
H. pylori ingested (childhood, fecal-oral)
↓
Urease + flagella → survives gastric acid, penetrates mucus
↓
Adhesins (BabA, SabA) → colonizes gastric epithelium
↓
T4SS injects CagA, PG, HBP → NF-κB → IL-8 → neutrophils
VacA → vacuolation, apoptosis
↓
Chronic active gastritis
/ \
Antral Corpus + Antrum
(hyperchlorhydria) (atrophy, hypochlorhydria)
↓ ↓
DU / GU Intestinal metaplasia
↓
Dysplasia → Adenocarcinoma
MALToma
Sources:
- Yamada's Textbook of Gastroenterology, 7th ed., Ch. 49 & 51
- Robbins & Cotran Pathologic Basis of Disease (Robbins), Ch. 17
- Jawetz, Melnick & Adelberg's Medical Microbiology, 28th ed.
- Sherris & Ryan's Medical Microbiology, 8th ed.
- Maingot's Abdominal Operations
This is a shared conversation. Sign in to Orris to start your own chat.