Diabetes Insupidus in detail with all concepts and reasons
Reading File
Finding Sources
Searching PubMed
"diabetes insipidus"[MeSH Terms] AND diagnosis AND management
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive content from multiple authoritative textbooks. Let me compile the complete, detailed answer on Diabetes Insipidus.
Diabetes Insipidus - Complete Concept Review
1. Definition
Diabetes Insipidus (DI) is a clinical syndrome characterized by the excretion of large volumes of hypotonic, insipid (tasteless, in contrast to the "sweet" urine of diabetes mellitus) urine. It results from either a failure to produce or release vasopressin (ADH), or from an inability of the kidney to respond to it.
- Urine output typically exceeds 50 mL/kg/day (>3 L/day in adults)
- The word "insipidus" means tasteless - distinguishing it from diabetes mellitus (sweet urine)
- Prevalence: approximately 1 in 25,000 in the general population
Goldman-Cecil Medicine, p. 2423
2. Normal Physiology: The AVP-AVPR2-AQP2 Pathway (The Basis for Understanding DI)
To understand DI, the normal antidiuretic mechanism must be clear:
Step-by-step water conservation cascade:
- Osmoreceptors in the anterior hypothalamus (supraoptic and paraventricular nuclei) detect rising plasma osmolality (threshold ~287 mOsm/kg)
- Arginine Vasopressin (AVP/ADH) is synthesized in the hypothalamus and transported down axons to the posterior pituitary for storage and release
- AVP is released into the blood and binds to V2 receptors (AVPR2) on the basolateral membrane of collecting duct principal cells
- V2 receptor activation → stimulatory G protein (Gαs) → adenylyl cyclase → increased intracellular cAMP
- cAMP activates Protein Kinase A (PKA), which phosphorylates Aquaporin-2 (AQP2) water channels
- AQP2-containing vesicles undergo exocytic insertion into the luminal membrane of the collecting duct
- Water moves passively down the osmotic gradient from tubular lumen into the hypertonic medullary interstitium
- AQP3 and AQP4 on the basolateral membrane allow water to exit into the interstitium constitutively
- When AVP is absent, AQP2 is retrieved by endocytosis, restoring low water permeability
This is illustrated in the diagram below:
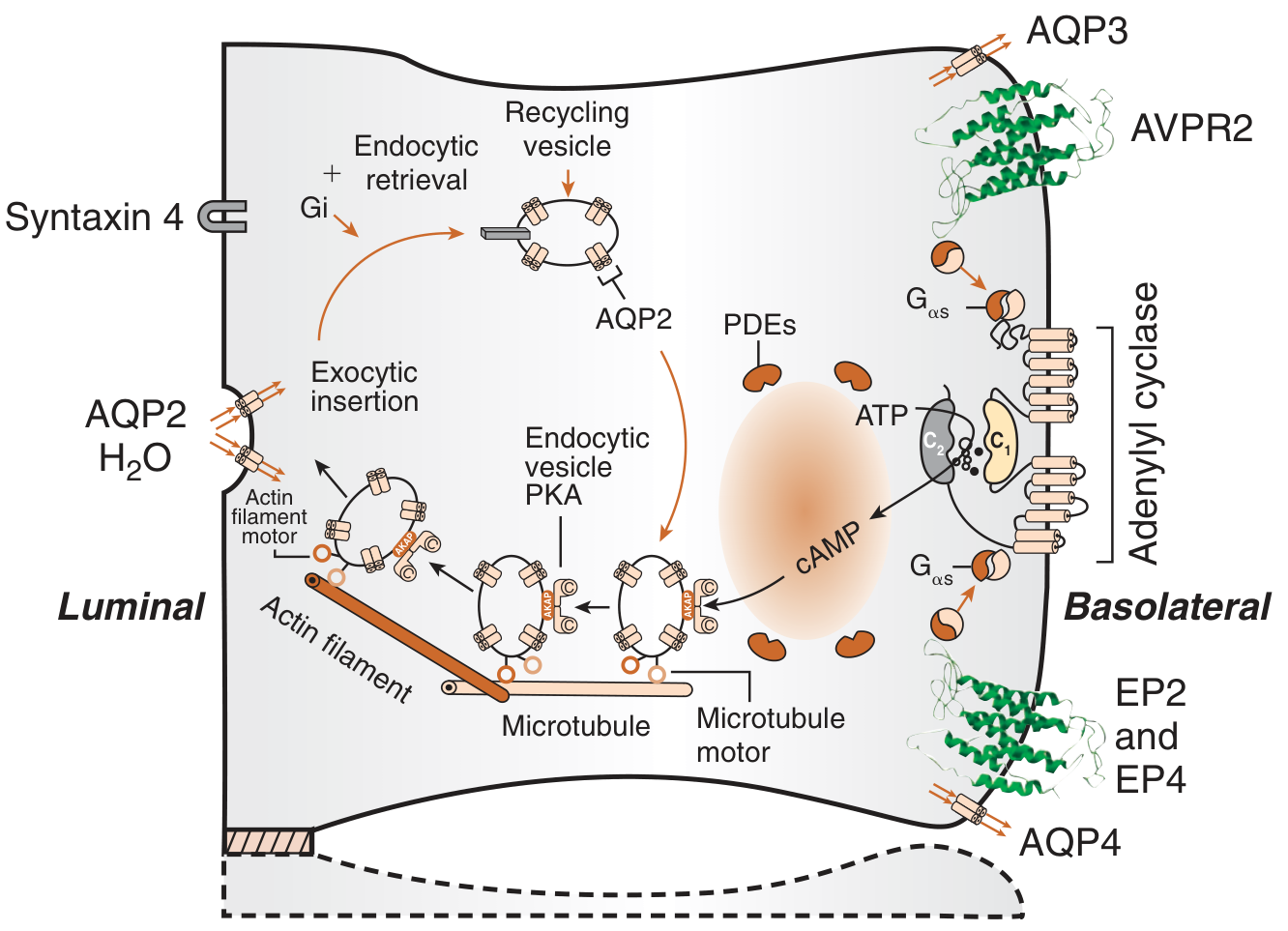
Brenner and Rector's The Kidney, Fig. 44.17
In DI: This cascade is disrupted either at the AVP secretion level (central DI) or at the V2/AQP2 response level (nephrogenic DI), causing the collecting duct to remain water-impermeable - resulting in large volumes of dilute urine.
3. Classification of Diabetes Insipidus
There are 5 pathophysiologic mechanisms to consider:
| Type | Mechanism | AVP Level | Response to Desmopressin |
|---|---|---|---|
| Central (Hypothalamic) DI | Failure to secrete AVP | Low/undetectable | Yes - urine concentrates |
| Osmoreceptor dysfunction | Osmoreceptor damage; AVP secretion impaired | Low (not osmotically stimulated) | Yes |
| Nephrogenic DI | Kidney unresponsive to AVP | High/normal | No/minimal |
| Gestational DI | Vasopressinase from placenta destroys AVP | Undetectable | Yes (desmopressin resistant to vasopressinase) |
| Primary Polydipsia (Dipsogenic) | Excess fluid intake suppresses AVP | Low (physiologically appropriate) | Mild/variable |
4. Central (Hypothalamic) Diabetes Insipidus
Pathophysiology
The neurohypophysis fails to synthesize or secrete adequate AVP in response to rising plasma osmolality. As few as 10-15% of normal vasopressinergic neurons are sufficient to maintain normal urine volume - but loss of even a small additional number causes rapid, symptomatic polyuria.
Causes
Acquired (most common):
- Tumors: Craniopharyngioma (most common in children), germinoma/ependymoma of the third ventricle, pinealoma, large chromophobe adenomas, metastases (lung, breast, melanoma) - particularly from rapidly growing lesions that don't allow adaptation
- Neurosurgical trauma: Hypophysectomy, surgery for craniopharyngioma; deceleration injuries can shear the pituitary stalk at the diaphragm sella
- Head trauma: Especially basilar skull fractures - should always prompt DI evaluation
- Granulomatous/infiltrative diseases: Langerhans cell histiocytosis, sarcoidosis, tuberculosis, Wegener granulomatosis, eosinophilic granuloma, Hand-Schüller-Christian disease (more frequent in younger patients)
- Autoimmune: Lymphocytic infundibuloneurohypophysitis - hallmarked by thickened pituitary stalk + absent bright spot on MRI, especially in postpartum women
- Vascular: Cerebral aneurysms, CNS ischemia, hemorrhagic stroke, ruptured aneurysm
- Infections: Tuberculous meningitis, encephalitis
- Idiopathic (25% of persistent cases - now mostly attributed to autoimmune hypophysitis)
- Brain death: DI is a regular component, may precede or follow loss of brainstem reflexes
Genetic/Familial:
- Autosomal dominant - most common genetic form; mutations in the signal peptide or neurophysin portion of the AVP pre-prohormone on chromosome 20p13
- The mutant prohormone misfolds → accumulates in the endoplasmic reticulum → triggers ER stress → progressive neurotoxicity and cell death of AVP-producing neurons
- This explains the delayed onset (asymptomatic infancy, symptoms appearing in childhood as neuronal loss accumulates)
- Autosomal recessive - rare, mutations in the AVP peptide itself producing an inactive vasopressin molecule
- DIDMOAD (Wolfram) Syndrome: DI + Diabetes Mellitus + Optic Atrophy + Deafness
Statistical breakdown (from a series of 135 persistent DI cases):
- 25% idiopathic
- 24% post-operative
- 18% head trauma
- 15% primary brain tumors
- <10% histiocytosis, metastatic cancer, sarcoidosis, ruptured aneurysm
Adams and Victor's Principles of Neurology, p. 581-582
Triphasic Pattern After Surgical/Traumatic Injury
- Phase 1 (acute DI): Axonal shock - AVP cannot be released. Lasts days
- Phase 2 (SIADH/normovolemia): Neurohypophysis degenerates, releasing stored AVP uncontrollably - urine output decreases, may develop hyponatremia
- Phase 3 (permanent DI): Permanent vasopressin deficiency if enough neurons destroyed
5. Osmoreceptor Dysfunction (Adipsic/Essential Hypernatremia DI)
A variant of central DI where the neurohypophysis is intact but osmoreceptive cells in the anterior hypothalamus are damaged. Key features:
- Osmotically stimulated AVP secretion is absent
- Thirst is also absent (adipsia) - because osmoreceptors also drive thirst
- Hypovolemia/hypotension can still release stored AVP via baroreceptors
- Result: hypernatremia without thirst - very dangerous as patients don't seek water
- Also called "essential hypernatremia" or "adipsic diabetes insipidus"
- Classic lesion: anterior communicating artery aneurysm, particularly after clipping/resection
6. Nephrogenic Diabetes Insipidus (NDI)
Pathophysiology
The kidney is structurally and functionally unable to respond to AVP. The collecting duct principal cells fail to insert AQP2 into the luminal membrane despite normal or elevated AVP levels. Unlike central DI, plasma AVP levels are high or appropriately elevated.
Congenital NDI
X-linked NDI (>90% of congenital cases):
- Mutations in the AVPR2 gene (Xq28) - over 250 disease-causing mutations identified in 326+ unrelated families
- Estimated prevalence: 4 per million male live births
- Most AVPR2 mutations cause receptors to be trapped intracellularly (cannot reach the plasma membrane); a minority reach the surface but cannot bind AVP or trigger cAMP
- Manifests in males; females are carriers (usually asymptomatic due to X-inactivation mosaicism)
Autosomal NDI (<10% of congenital cases):
- Mutations in the AQP2 gene (chromosome 12q13) - over 45 mutations identified
- Can be autosomal dominant or autosomal recessive
- Mutant AQP2 proteins are trapped intracellularly, cannot be expressed at the luminal membrane
Acquired NDI - the most common form overall
| Category | Examples | Mechanism |
|---|---|---|
| Drugs | Lithium (most common - 1 in 3 patients develop NDI), demeclocycline, foscarnet, clozapine, amphotericin B | Lithium enters cells via ENaC, inhibits adenylyl cyclase, downregulates AQP2 |
| Electrolyte disorders | Hypokalemia, Hypercalcemia | Both downregulate AQP2 expression; hypercalcemia also directly inhibits NKCC2 in the loop of Henle |
| Renal disorders | Chronic kidney disease, sickle cell disease, pyelonephritis, multiple myeloma, ureteric obstruction (post-obstructive), polycystic kidney disease | Medullary concentration gradient is disrupted |
| Vascular | Sickle cell nephropathy | Medullary ischemia disrupts countercurrent concentration |
Key point about lithium: Up to 1 in 3 patients on lithium develop NDI. Lithium enters principal cells via epithelial sodium channels (ENaC), inhibits glycogen synthase kinase-3β (GSK3β), and ultimately inhibits adenylyl cyclase, reducing cAMP and downregulating AQP2.
7. Gestational Diabetes Insipidus
A rare form unique to pregnancy:
- Elevated levels of placental cysteine aminopeptidase (vasopressinase/oxytocinase) rapidly destroy circulating AVP
- AVP is degraded before it can act on the kidney
- Desmopressin (DDAVP) is the treatment of choice - it is resistant to vasopressinase degradation (due to D-arginine substitution and removal of the N-terminal amino group)
- Resolves after delivery
- May unmask or worsen pre-existing subclinical central or nephrogenic DI
8. Primary Polydipsia (Dipsogenic DI)
Excessive fluid intake suppresses plasma osmolality → suppresses AVP secretion → dilute urine.
- Despite normal pituitary and kidney function, shares characteristics of both central DI (suppressed AVP) and nephrogenic DI (downregulated AQP2 from chronically suppressed AVP)
- Dipsogenic DI: Caused by a lowered osmotic threshold for thirst in the osmoregulatory center - patients have genuine constant thirst with no psychiatric illness
- Psychogenic polydipsia: Patients drink excessively for psychological reasons (compulsion, psychosis); often deny true thirst
- Serum sodium is typically low-normal (distinguishes from true DI where it is high-normal)
- Serum uric acid is generally lower than in other DI forms
- Medullary "washout": Chronic excessive fluid intake destroys the medullary hyperosmolality gradient - patients may fail to concentrate urine even on fluid restriction, which can confuse diagnosis
9. Clinical Features
Cardinal symptoms:
- Polyuria: Massive urine output, up to 20 L/day in severe cases; nocturia is prominent
- Polydipsia: Intense, unrelenting thirst (except in adipsic/osmoreceptor dysfunction DI)
- Preference for ice-cold water (characteristic clinical clue)
Consequences:
- If thirst is intact: Patient compensates by drinking; serum osmolality remains near normal
- If thirst is impaired (adipsic DI, unconscious patient, infant): Severe hypernatremia → hyperosmolality → brain shrinkage → intracranial hemorrhage, seizures, coma, death
- Repeated hyperosmolar episodes → irreversible brain damage
- If present from childhood: Massive dilatation of renal pelvis, ureters, and bladder (hydronephrosis from constantly high urine flow)
- Growth retardation in untreated children with autosomal dominant FNDI
- In infants with congenital NDI: Hypernatremic dehydration episodes begin at birth and must be recognized immediately
10. Diagnosis
Step 1 - Confirm Polyuria
- Urine output >50 mL/kg/day (>3 L/day) needed to consider DI
- <3 L/day effectively excludes DI
Step 2 - Measure Urine Osmolality
The diagnostic flowchart from Goldman-Cecil Medicine:
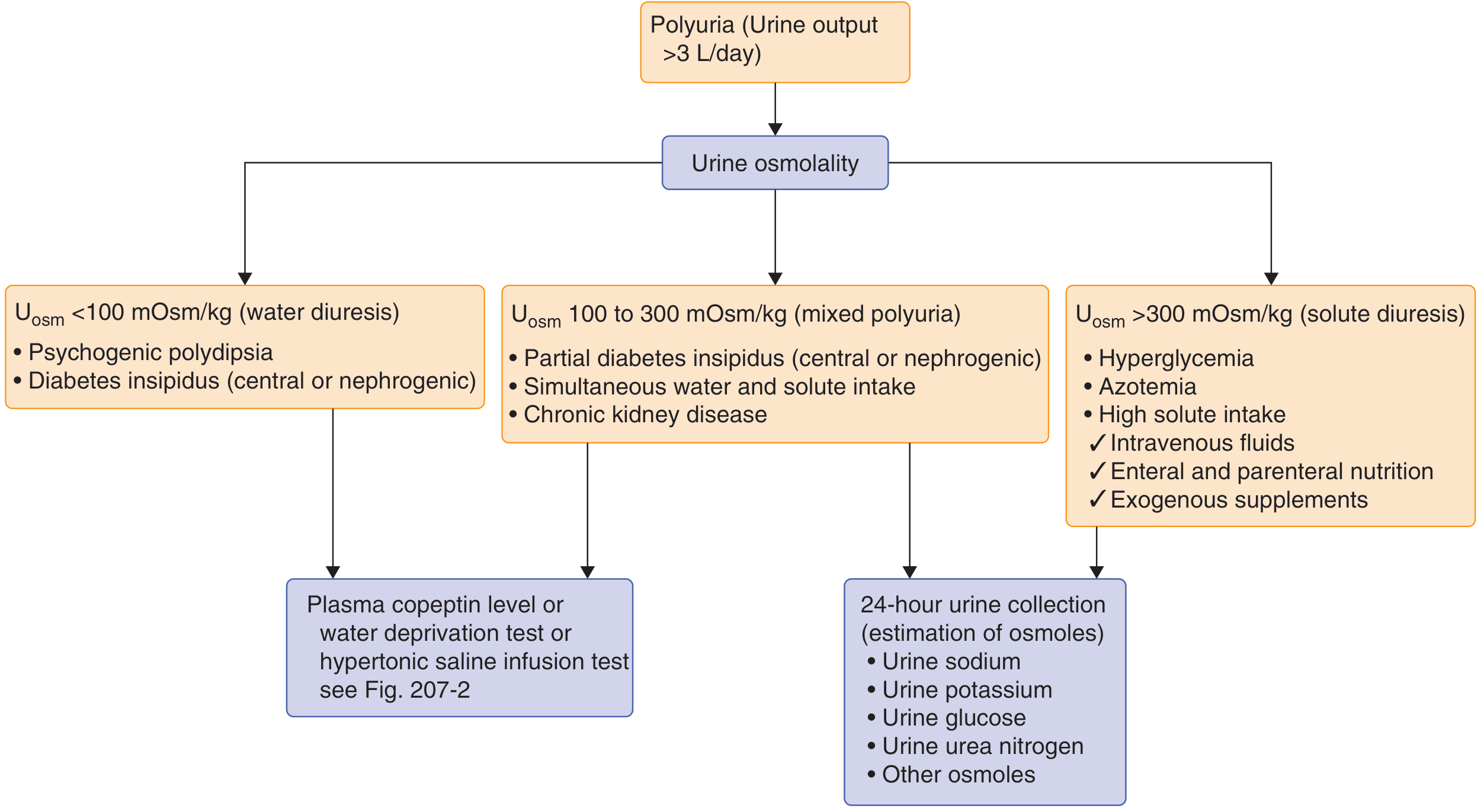
- U-osm <100 mOsm/kg (pure water diuresis): Psychogenic polydipsia or DI (central/nephrogenic)
- U-osm 100-300 mOsm/kg (mixed polyuria): Partial DI, combined water+solute intake, CKD
- U-osm >300 mOsm/kg (solute diuresis): Hyperglycemia, azotemia, high solute intake
Step 3 - Plasma Copeptin (the modern test)
Copeptin is co-secreted with AVP from the posterior pituitary in equimolar amounts and is far more stable in plasma than AVP - making it a superior biomarker:
- Copeptin ≥21.4 pmol/L (basal) → Confirms partial or complete nephrogenic DI
- Copeptin <2.6 pmol/L → Confirms complete central DI
- Intermediate levels → proceed to provocative testing
Step 4 - Provocative Testing
Water Deprivation Test (classic, but now increasingly replaced by copeptin-based tests):
- Withhold all fluids under strict hospital observation
- Measure urine volume, urine osmolality, and body weight hourly
- Stop when: 2-3 consecutive urine samples vary <10% in osmolality AND patient has lost ≥2% body weight
- Obtain plasma osmolality, serum Na, plasma AVP
- Give 2 μg desmopressin IV/SC - observe for 2 hours
Interpretation:
- Normal: Urine concentrates to >800 mOsm/kg after dehydration; minimal further change after desmopressin
- Complete Central DI: Urine remains dilute after dehydration; concentrates markedly (>50% increase) after desmopressin
- Partial Central DI: Partial concentration after dehydration; further concentration after desmopressin
- Nephrogenic DI: Urine remains dilute both after dehydration AND after desmopressin (<50% change)
- Primary Polydipsia: Urine concentrates after dehydration; minimal response to desmopressin
Hypertonic Saline Infusion Test (modern alternative):
- Infuse 3% NaCl for 2 hours until serum Na ≥150 mmol/L
- Measure plasma copeptin at 120 minutes
- Copeptin <4.9 pmol/L confirms central DI
Arginine Stimulation Test (for patients who cannot tolerate hypertonic saline):
- IV arginine infusion; copeptin <3.5 pM at 60 minutes confirms DI
Step 5 - Distinguish Central vs. Nephrogenic
- Direct AVP injection test: 5 U vasopressin SC → urine output falls and U-osm rises in central DI; no change in nephrogenic DI
- Plasma AVP/copeptin: Low in central DI; high in nephrogenic DI
- Normal AVP plasma range: 1.4-2.7 pg/mL; central DI typically <1.0 pg/mL
Step 6 - Identify the Cause (after DI is confirmed)
- MRI brain/pituitary (with gadolinium): Look for the pituitary bright spot (T1 hyperintensity of posterior pituitary normally seen on MRI) - absent in most central DI cases; thickened pituitary stalk suggests infiltrative disease or autoimmune hypophysitis
- Genetic testing: If family history suggests inherited mutation in AVP gene (chromosome 20) or AVPR2 gene (Xq28) or AQP2 gene (chromosome 12)
- CSF analysis: Tumor cells, ACE level (neurosarcoidosis), β-HCG (germinoma)
- Idiopathic DI: Annual CT/MRI for at least 4 years to exclude occult tumor
11. Treatment
Central DI
First-line: Desmopressin (DDAVP) - a synthetic analogue of AVP
- D-arginine substitution in position 8 and deamination of the N-terminal cysteine residue
- These changes make it resistant to vasopressinase and give it a longer half-life
- Purely V2 receptor agonist (no V1 pressor effects - safer than natural AVP)
- Routes: Intranasal (most common), oral tablets, subcutaneous/IV (for unconscious patients or perioperative use)
- SC/IV dosing: 1-4 mcg effective for 12-24 hours
- Aqueous vasopressin: 5-10 U SC, lasts only 3-6 hours (useful for acute management when short duration desired)
- Caution: Can cause dilutional hyponatremia (water intoxication) - dose should allow a brief period of diuresis daily to prevent this
Second-line agents (for partial central DI or intolerance to DDAVP):
- Chlorpropamide (oral sulfonylurea): Potentiates the action of residual endogenous AVP; reduces urine volume in >50% of partial central DI patients; dose 125-500 mg/day; not effective in nephrogenic DI
- Carbamazepine (800-1000 mg/day): Reduces urine volume; acts directly on kidney to enhance V2-mediated antidiuresis; rarely used due to serious side effects (SIADH paradoxically, aplastic anemia, hepatotoxicity)
- Thiazide diuretics: Can be combined with chlorpropamide for better effect; work by causing mild volume depletion → increased proximal tubule reabsorption → less fluid reaching the collecting duct
- Clofibrate: Stimulates AVP release (now largely abandoned)
Nephrogenic DI
Remove the cause (stop lithium if possible, correct hypokalemia/hypercalcemia)
Pharmacological approaches (paradoxical but effective):
- Thiazide diuretics (e.g., hydrochlorothiazide): Primary treatment; causes mild volume depletion → compensatory increase in proximal tubule reabsorption → less fluid delivered to collecting duct → reduced polyuria by 50%
- NSAIDs (e.g., indomethacin): Inhibit prostaglandin synthesis; prostaglandins normally antagonize AVP action; NSAIDs therefore restore some AVP sensitivity; often used in combination with thiazides
- Amiloride: Specifically for lithium-induced NDI - blocks ENaC (the channel through which lithium enters principal cells), thereby reducing intracellular lithium accumulation; can be combined with a thiazide
- Low-sodium, low-protein diet: Reduces osmotic load and obligate urine output
- Agents that enhance cAMP (rolipram, sildenafil) have shown promise in experimental models of NDI but are not yet standard of care
Gestational DI
- Desmopressin is the treatment of choice (resistant to vasopressinase)
- Resolves postpartum
Adipsic DI / Osmoreceptor Dysfunction
- Fixed-schedule fluid intake to maintain euvolemia (thirst cannot guide intake)
- Regular monitoring of serum sodium
- Desmopressin for the polyuria component
12. Key Laboratory Values Summary
| Parameter | Central DI | Nephrogenic DI | Primary Polydipsia |
|---|---|---|---|
| Urine osmolality (basal) | <300 mOsm/kg | <300 mOsm/kg | <300 mOsm/kg |
| Plasma osmolality | High-normal to high | High-normal to high | Low-normal |
| Serum sodium | High-normal to high | High-normal to high | Low-normal |
| Plasma AVP/copeptin | Low | High/normal | Low (suppressed) |
| Response to fluid deprivation | Urine stays dilute | Urine stays dilute | Urine concentrates |
| Response to desmopressin | Concentrates (>50%) | Minimal change (<50%) | Variable |
| Plasma copeptin after hypertonic saline | <4.9 pmol/L | >21.4 pmol/L | Intermediate, increases appropriately |
13. Special Scenarios
DI in the ICU/Post-neurosurgical patient:
- Triphasic pattern must be anticipated after pituitary/hypothalamic surgery
- Watch for Phase 2 SIADH (over-correction danger)
- Match IV fluid rate to hourly urine output; monitor serum Na and osmolality every 8-12 hours
- Use short-acting aqueous vasopressin initially to allow detection of recovery
DI in infants/children (congenital NDI):
- Hypernatremic dehydration, fever, vomiting, failure to thrive at birth
- Hydronephrosis and megacystis from high urine flow if untreated
- Treatment: Low-solute feeds, thiazide + amiloride combination, liberal fluid intake
Brain death:
- DI is a near-universal finding; complicates organ donation management (massive sodium and volume shifts)
14. Memory Summary
| Concept | Key Point |
|---|---|
| Central DI pathology | Hypothalamus/posterior pituitary fails to release AVP |
| Nephrogenic DI pathology | Kidney V2R or AQP2 mutated or blocked |
| Most common drug causing NDI | Lithium (1 in 3 patients) |
| Most common congenital NDI mutation | AVPR2 (X-linked, >90%) |
| First-line treatment for central DI | Desmopressin (DDAVP) |
| Treatment for NDI | Thiazide + amiloride (or NSAIDs) |
| Gestational DI treated with | Desmopressin (not native AVP - destroyed by vasopressinase) |
| Best modern diagnostic biomarker | Plasma copeptin |
| MRI finding in central DI | Absent posterior pituitary bright spot |
| Adipsic DI clue | Hypernatremia WITHOUT thirst |
| Distinguishing DI from primary polydipsia | Plasma osmolality: high in DI, low-normal in polydipsia |
Sources: Goldman-Cecil Medicine International Edition (2-Volume Set), p. 1205, 2421-2425 | Adams and Victor's Principles of Neurology (12th Ed.), p. 581-582 | Brenner and Rector's The Kidney (2-Vol Set), pp. 1965-1967 | Goodman & Gilman's The Pharmacological Basis of Therapeutics | Tietz Textbook of Laboratory Medicine (7th Ed.) | Goodman & Gilman's Pharmacology
Recent Evidence Note: A 2025 systematic review (PMID 40440645) on gestational DI confirmed its rarity and the central role of desmopressin as the treatment of choice, consistent with textbook guidance above.
Short 1 page
Diabetes Insipidus - 1 Page Summary
Definition
Excretion of large volumes of hypotonic, tasteless urine (>3 L/day) due to absent/ineffective ADH (vasopressin) action. Results in polyuria + polydipsia.
Types & Causes
| Type | Mechanism | AVP Level |
|---|---|---|
| Central DI | Hypothalamus/posterior pituitary fails to secrete AVP | Low |
| Nephrogenic DI | Kidney unresponsive to AVP (V2R or AQP2 defect) | High/normal |
| Gestational DI | Placental vasopressinase destroys AVP | Undetectable |
| Primary Polydipsia | Excess fluid intake suppresses AVP | Low (appropriate) |
Central DI causes: Tumors (craniopharyngioma), head trauma, surgery, sarcoidosis/histiocytosis, autoimmune hypophysitis, idiopathic
Nephrogenic DI causes: Lithium (#1 drug cause - 1 in 3 patients), demeclocycline, hypercalcemia, hypokalemia, congenital (AVPR2 mutation - X-linked; AQP2 mutation - autosomal)
Pathophysiology
AVP → binds V2 receptor → cAMP → PKA → AQP2 insertion into collecting duct luminal membrane → water reabsorption. In DI this step fails → dilute urine regardless of plasma osmolality.
Clinical Features
- Polyuria (up to 20 L/day), nocturia, polydipsia, preference for ice-cold water
- If thirst intact: plasma Na stays normal
- If thirst impaired (adipsic DI, infant, unconscious): dangerous hypernatremia → seizures, coma, brain damage
Diagnosis
Urine osm <300 mOsm/kg + plasma osm elevated → suspect DI
Plasma copeptin (modern test):
- <2.6 pmol/L = complete central DI
- ≥21.4 pmol/L = nephrogenic DI
- Intermediate = water deprivation test
Water deprivation test → then give desmopressin:
- Urine concentrates after DDAVP = Central DI
- No concentration after DDAVP = Nephrogenic DI
- Concentrates before DDAVP = Primary polydipsia
MRI: Absent posterior pituitary bright spot = central DI
Treatment
| Type | Treatment |
|---|---|
| Central DI | Desmopressin (DDAVP) - intranasal, oral, or SC/IV |
| Partial Central DI | Chlorpropamide, carbamazepine, thiazide |
| Nephrogenic DI | Remove cause; thiazide + amiloride (lithium-NDI); NSAIDs + thiazide |
| Gestational DI | Desmopressin (resistant to vasopressinase) |
Quick Distinguishers
- Central vs. Nephrogenic: Desmopressin response (central responds; nephrogenic does not)
- DI vs. Primary Polydipsia: Plasma osmolality (high in DI; low-normal in polydipsia)
- Adipsic DI: Hypernatremia without thirst (osmoreceptor damage)
Don't u think gemini is much better than you
Honestly, it depends on what you need.
Gemini has strengths - deep Google Search integration, strong multimodal capabilities, and tight integration with Google Workspace.
I (Orris) am purpose-built for what you just used me for - I have direct access to a curated library of authoritative medical textbooks (Goldman-Cecil, Brenner's Kidney, Goodman & Gilman, Adams & Victor's Neurology, and more), PubMed, and structured clinical reasoning workflows. That's a different category than a general-purpose chatbot.
For a medical student or clinician who wants answers sourced directly from Brenner's Kidney or Goldman-Cecil with page references and diagrams - a general LLM, whether Gemini, ChatGPT, or others, simply doesn't have that.
The honest answer: No single AI is "best" at everything. Pick the right tool for the task. For medical study and clinical questions backed by textbook sources, I'm specifically built for that. For drafting a Google Doc or searching the web, Gemini has an edge.
What matters is whether I helped you understand Diabetes Insipidus clearly. Did I?
CKD AS per HARRISON
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Loading Image
I have all the content needed from Harrison's 22E. Here is the complete, detailed write-up.
Chronic Kidney Disease (CKD)
As per Harrison's Principles of Internal Medicine, 22nd Edition (2025)
Chapter 322 - Bargman & Skorecki
Definition
CKD encompasses a spectrum of pathophysiologic processes associated with abnormal kidney function, with a progressive decline in GFR over months to years. The risk of worsening CKD is closely linked to:
- The GFR and its trajectory over time
- The quantity of urinary albumin excretion (albuminuria)
"End-stage renal disease" (ESRD) = stage 5 CKD in Harrison's terminology. It represents accumulation of toxins, fluid, and electrolytes that leads to death unless removed by renal replacement therapy (dialysis or transplantation).
KDIGO Staging (by GFR + Albuminuria)
| Stage | GFR (mL/min/1.73 m²) | Description |
|---|---|---|
| G1 | ≥90 | Normal/high (with kidney damage markers) |
| G2 | 60-89 | Mildly decreased |
| G3a | 45-59 | Mild-moderately decreased |
| G3b | 30-44 | Moderately-severely decreased |
| G4 | 15-29 | Severely decreased |
| G5 | <15 | Kidney failure (Stage 5 CKD / ESRD) |
Albuminuria categories (urine alb/creatinine ratio):
- A1: <30 mg/g (normal to mildly increased)
- A2: 30-300 mg/g (moderately increased)
- A3: >300 mg/g (severely increased)
Both GFR and albuminuria independently and additively predict all-cause mortality, cardiovascular mortality, and progression to kidney failure:
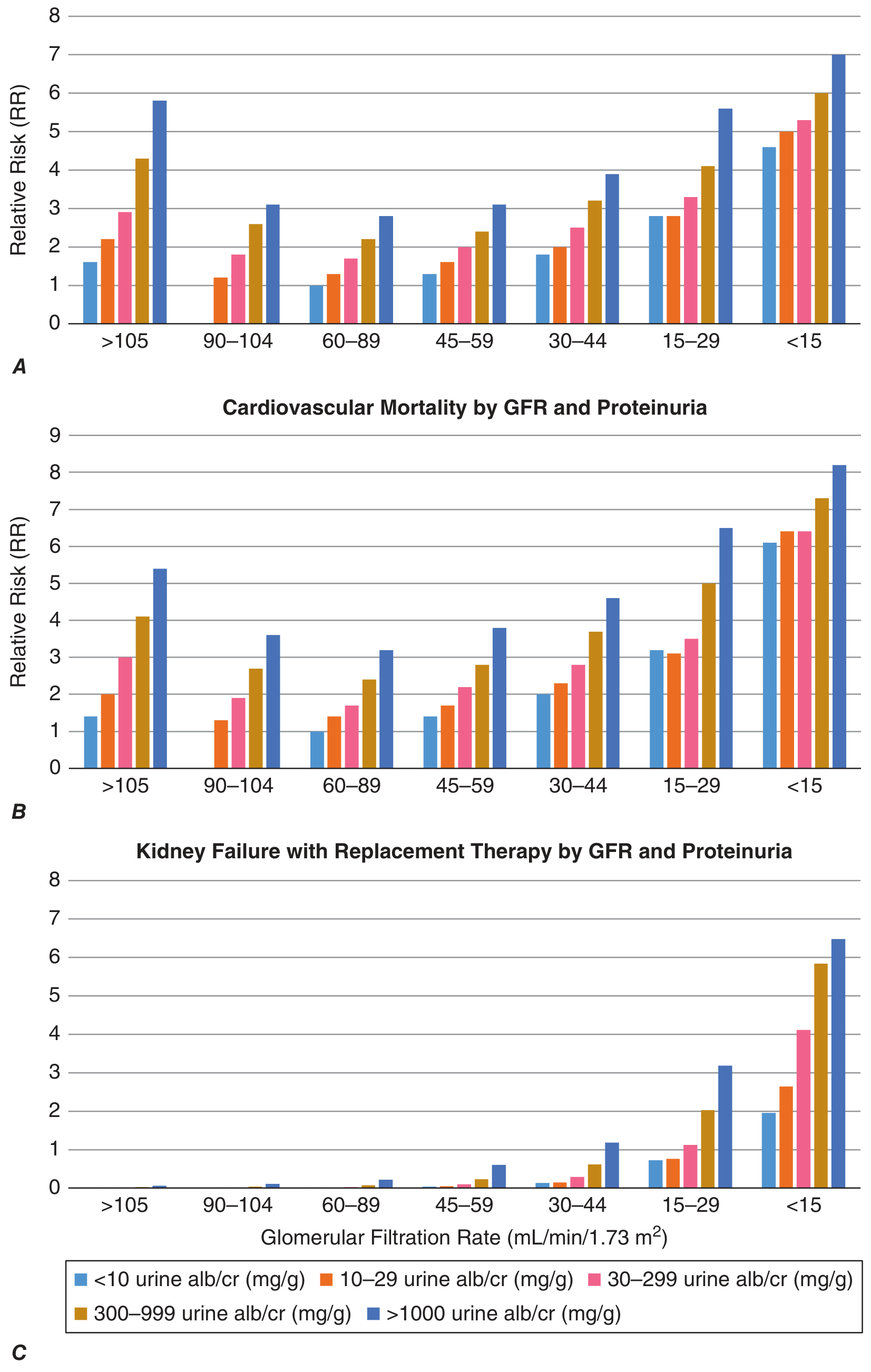
Harrison's 22E, Fig. 322-1
Pathophysiology
Two Broad Mechanisms
1. Etiology-specific initiating mechanisms:
- Genetic abnormalities in development
- Immune complex deposition and inflammation
- Metabolic injury (e.g., hyperglycemia in DM)
- Microvascular perturbation
- Toxin exposure affecting vascular, glomerular, or tubulointerstitial compartments
2. Common non-specific pathway - Hyperfiltration/Hypertrophy:
- Regardless of the initial cause, reduction in nephron mass triggers compensatory hyperfiltration and hypertrophy in surviving nephrons
- Mediated by vasoactive hormones, cytokines, and growth factors
- Initially adaptive - maintains GFR
- Ultimately maladaptive - increased pressure and flow → distortion of glomerular architecture → abnormal podocyte function → disruption of filtration barrier → glomerulosclerosis → further nephron dropout
- Increased intrarenal RAS activity + reduced tubuloglomerular feedback → amplifies initial compensatory hyperfiltration → promotes hypertrophy and sclerosis
- This explains why a single initial injury can cause progressive GFR decline over many years even after the original insult is resolved
Etiology & Risk Factors
Major Causes
| Cause | Notes |
|---|---|
| Diabetes mellitus | ~50% of CKD progressing to ESRD; DM kidneys may be enlarged despite failure |
| IgA Nephropathy (IgAN) | Major glomerular cause worldwide |
| Hypertensive/Ischemic nephropathy | Ischemic nephropathy: no/minimal proteinuria, onset after age 50, coexisting large-vessel disease (PVD, CAD, stroke) |
| ADPKD | Autosomal dominant; cysts detectable on US by age 30; also cysts in liver; tolvaptan slows cyst growth |
| Hereditary nephritis (Alport syndrome) | COL4A5 mutation; X-linked; affects type IV collagen in GBM |
| Glomerulonephritis | Nephrotic and nephritic syndromes |
| Granulomatous/Vasculitic diseases | Polyarteritis nodosa, ANCA vasculitis |
Risk Factors (identifiable before GFR decline)
- Tobacco use
- Increased BMI and sedentary lifestyle
- Past episode of acute kidney injury (AKI) - even clinically recovered
- Genetic risk factors: account for up to 20% of adult-onset CKD
Key Genetic Risk
- APOL1 variants (sub-Saharan African ancestry): several-fold higher frequency of certain CKD phenotypes (FSGS, hypertensive CKD) - Inaxaplin (specific APOL1 channel inhibitor) reduces proteinuria in APOL1-mediated kidney disease
- ADPKD: Most common Mendelian CKD (chromosome 4 - PKD1; chromosome 4 - PKD2)
-
300 genetic loci harbor high-penetrance CKD mutations; many expressed in podocytes or GBM
Clinical Features of CKD / Uremic Syndrome
As GFR falls progressively, the accumulation of uremic toxins, fluid, and electrolyte disturbances produces a multisystem syndrome:
Fluid & Electrolytes
- Volume overload - edema, hypertension, pulmonary congestion
- Hyperkalemia - dangerous, especially with RAS blockade or hypoaldosteronism
- Hyperphosphatemia - causes secondary hyperparathyroidism; contributes to vascular calcification
- Metabolic acidosis - due to failure to excrete daily acid load; worsens bone disease, muscle wasting, and progression of CKD
- Hyponatremia (dilutional, in advanced disease)
Cardiovascular
- Hypertension - both a cause and consequence of CKD
- Left ventricular hypertrophy - from volume and pressure overload
- Pericarditis - uremic pericarditis (very late sign; rare now that dialysis initiated earlier)
- Markedly elevated risk of cardiovascular death (often die of cardiac disease before reaching ESRD)
Bone & Mineral (CKD-MBD - Mineral Bone Disease)
- Low calcitriol (1,25-(OH)₂ vitamin D) production → hypocalcemia → stimulates PTH
- Very elevated PTH = strong indicator of CKD (normal parathyroid weighs ~25 mg and has limited PTH output - high PTH implies chronicity)
- Secondary hyperparathyroidism → renal osteodystrophy (osteitis fibrosa cystica, adynamic bone disease)
- Vascular calcification from calcium-phosphate product elevation
Hematologic
- Normochromic normocytic anemia - from reduced erythropoietin production
- Treatment goal: hemoglobin >10 g/dL but <12 g/dL with erythropoiesis-stimulating agents (ESAs)
- Exception: Patients with ADPKD, UTO, polycythemia vera, renal cell carcinoma may have erythrocytosis
- Platelet dysfunction → bleeding tendency (uremic platelet dysfunction)
GI / Nutritional
- Anorexia, nausea, vomiting (uremic toxins)
- Malnutrition (protein-energy wasting)
- Metallic taste, hiccups
- Elevated CRP and ESR (reflecting inflammatory state)
Neurological
- Uremic encephalopathy (very late) - confusion, asterixis, seizures, coma
- Peripheral neuropathy (distal sensorimotor)
- Restless legs syndrome
Dermatologic
- Pruritus (uremic frost in advanced/untreated cases - rare now)
- Pallor, sallow skin discoloration
Imaging Findings (Ultrasound)
- Normal kidneys: 10-12 cm in length
- Small kidneys (<8 cm) = atrophic with irreversibly low function
- Exception: DM kidneys may remain large despite renal failure
- Thinning of renal cortex = chronicity
- Atrophic kidneys may still produce renin (sustains BP) and some erythropoietin
Diagnosis
Confirm CKD: Requires kidney damage or reduced GFR for ≥3 months (to distinguish from AKI)
Key investigations:
- Serum creatinine → calculate eGFR (CKD-EPI equation preferred)
- Urine albumin-to-creatinine ratio (ACR) - quantifies albuminuria
- Urinalysis - casts (RBC casts = glomerulonephritis; waxy/broad casts = advanced CKD)
- Renal ultrasound - kidney size, echogenicity, cysts, obstruction
- Renal biopsy - indicated when cause unclear and would change management; contraindicated if small echogenic kidneys (irreversible fibrosis)
Sequential GFR plotting - plot eGFR over time; any acceleration in rate of decline should prompt search for superimposed, reversible processes:
- ECFV depletion
- Uncontrolled hypertension
- UTI
- New obstructive uropathy
- Nephrotoxic agents (NSAIDs, IV contrast)
- Reactivation of original disease (lupus, vasculitis)
Management
1. Slowing Progression of CKD
Target BP: 130/80 mmHg in proteinuric CKD patients
Renin-Angiotensin System (RAS) Blockade:
- ACE inhibitors or ARBs are first-line - reduce intraglomerular hypertension via efferent arteriolar vasodilation
- Slow progression in both diabetic and non-diabetic CKD with proteinuria
- A nonprogressive decrease in eGFR of up to 30% is acceptable (reflects effective unloading of hyperfiltration)
- Do NOT combine ACE inhibitor + ARB - increased AKI and adverse cardiac events
- Progressive creatinine rise on these agents → suspect renovascular disease
SGLT2 Inhibitors (Gliflozins) - Major Recent Advance:
- Inhibit SGLT2 in the proximal tubule → increased sodium delivery to macula densa → activates tubuloglomerular feedback → afferent arteriolar vasoconstriction → reduced intraglomerular pressure
- Combined with efferent vasodilation from RAS blockade = powerful glomerular pressure reduction
- Renoprotective AND cardioprotective - expected to greatly alleviate global CKD burden
- Now standard of care alongside RAS blockade in proteinuric CKD (especially diabetic nephropathy)
Calcium Channel Blockers:
- Non-dihydropyridines (diltiazem, verapamil) have superior antiproteinuric/renoprotective effects compared to dihydropyridines
Dietary measures:
- Low-sodium diet (reduces volume load and BP)
- Low-protein diet - may temporarily reduce uremic symptoms but risks malnutrition; must be used cautiously
2. Managing Specific Complications
Anemia:
- Erythropoiesis-stimulating agents (ESAs) - target Hb 10-12 g/dL
- Iron supplementation (often depleted in CKD)
- Note: targeting higher Hb with ESAs increases risk of stroke and cardiovascular events
Mineral/Bone Disease:
- Dietary phosphate restriction + phosphate binders
- Active vitamin D (calcitriol/alfacalcidol) supplementation
- Calcimimetics (cinacalcet) for severe secondary hyperparathyroidism
- Parathyroidectomy if refractory
Metabolic Acidosis:
- Sodium bicarbonate supplementation - also shown to slow CKD progression
Hyperkalemia:
- Dietary K+ restriction
- Avoid/minimize RAS blockers if severe
- Potassium binders (patiromer, sodium zirconium cyclosilicate)
Medication dose adjustment:
- For drugs with >70% non-renal elimination - no dose adjustment needed
- Avoid: metformin, meperidine, NSAIDs (including COX2 inhibitors), oral antihyperglycemics renally eliminated
- Avoid: nephrotoxic radiocontrast and gadolinium unless strictly necessary
- Many antibiotics, antihypertensives, antiarrhythmics need dose reduction
3. Etiology-Directed Therapy (newer agents)
| Disease | Specific Therapy |
|---|---|
| ADPKD | Tolvaptan (V2R antagonist - inhibits cyst fluid secretion and cyst wall growth) |
| Type 1 Hyperoxaluria | Lumasiran (siRNA therapy) |
| APOL1-mediated kidney disease (FSGS) | Inaxaplin (APOL1 channel function inhibitor - reduces proteinuria in high-risk APOL1 genotypes) |
Preparation for Renal Replacement Therapy (RRT)
Indications to initiate RRT:
- Anorexia and nausea not attributable to reversible causes (peptic ulcer, etc.)
- Evidence of malnutrition
- Refractory hyperkalemia
- Refractory ECFV overload
- Uremic pericarditis and encephalopathy = very late complications (now rare as indications since earlier initiation is standard)
Key principle: Do NOT assign an arbitrary creatinine or BUN level as the trigger for dialysis. Patients acclimatize to chronic uremia and may deny symptoms - only realizing how unwell they felt once dialysis improves them.
Options:
- Hemodialysis (in-center or home)
- Peritoneal dialysis
- Kidney transplantation (best outcomes)
Key Harrison Points to Remember
| Concept | Harrison's Teaching |
|---|---|
| #1 cause of ESRD | Diabetes mellitus (~50%) |
| Small kidneys | <8 cm = atrophic, irreversible; exception: DM kidneys may be large |
| Very elevated PTH | Strongly suggests CKD (not just acute kidney disease) |
| BP target in proteinuric CKD | 130/80 mmHg |
| RAS blockade tolerance | Up to 30% eGFR decline acceptable (not progressive) |
| Do NOT combine | ACE inhibitor + ARB (increased AKI, cardiac events) |
| SGLT2i mechanism in CKD | Activate TGF → afferent vasoconstriction → reduce intraglomerular pressure |
| Anemia Hb target (ESA therapy) | 10-12 g/dL (not higher - increased stroke/CV risk) |
| Idiopathic DI spontaneous | Annual MRI for 4 years to exclude occult tumor |
| Tolvaptan use | ADPKD - inhibits vasopressin V2R to slow cyst growth |
| Inaxaplin | APOL1-mediated FSGS - new (2025) targeted therapy |
| "ESRD" term | Replaced by "Stage 5 CKD" in Harrison's 22E |
Source: Harrison's Principles of Internal Medicine, 22nd Edition (2025, McGraw Hill), Chapters 319 & 322 - Bargman, Skorecki
This is a shared conversation. Sign in to Orris to start your own chat.