multiple myeloma
Reading File
Finding Sources
Searching PubMed
"multiple myeloma"[MeSH Terms] AND treatment
Reading File
Reading File
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Loading Image
Searching the Web
multiple myeloma treatment guidelines 2025 daratumumab CAR-T
I have comprehensive content from multiple authoritative textbooks. Now I'll compile the full, detailed response.
Multiple Myeloma
Overview
Multiple myeloma (MM) is a malignant neoplasm of terminally differentiated B cells (plasma cells) that accumulate in the bone marrow, produce a monoclonal immunoglobulin (M protein), and cause systemic end-organ damage. It is one of the most common lymphoid malignancies, with approximately 30,000 new cases/year in the United States. The median age at diagnosis is 65-70 years; it is more common in males and, notably, occurs approximately twice as frequently in people of African descent compared to those of European descent.
- Robbins & Kumar Basic Pathology, p. 422
- Henry's Clinical Diagnosis and Management by Laboratory Methods, p. 758
Pathogenesis
MM arises from a post-germinal center plasma cell. Several key molecular events drive its development:
| Mechanism | Detail |
|---|---|
| IgH translocations | Chromosomal translocations fuse the IgH locus (chr 14) to oncogenes such as CCND1 (cyclin D1) and CCND3 (cyclin D3), dysregulating D cyclins and driving proliferation |
| IL-6 signaling | Interleukin-6 from bone marrow stromal fibroblasts and macrophages is the principal growth factor for myeloma cells |
| RANKL upregulation | Myeloma-derived factors upregulate RANKL on bone marrow stromal cells → osteoclast activation → bone resorption |
| Osteoblast suppression | Tumor-derived factors inhibit osteoblast function, creating the net lytic skeletal effect |
| MYC translocations | Appear late in disease, especially in aggressive/refractory cases |
Cytogenetics divide myeloma into two broad groups with prognostic significance:
- Hyperdiploid (trisomies of odd-numbered chromosomes): favorable prognosis
- Hypodiploid/non-hyperdiploid: adverse prognosis
Adverse markers: del(17p13) [TP53], t(4;14), t(14;16), t(14;20), del(13q14)
Favorable markers: t(11;14), t(6;14)
- Robbins & Kumar Basic Pathology, p. 422
- Henry's Clinical Diagnosis and Management, p. 758
M Protein Distribution
| Isotype | Frequency |
|---|---|
| IgG | ~60% |
| IgA | ~20-25% |
| Light chain only (κ or λ) | ~20% |
| IgD, IgE, IgM | Rare |
| Nonsecretory | ~1% |
Clinical Features - the "CRAB" Criteria
End-organ damage defines symptomatic myeloma requiring treatment:
| C | Hypercalcemia | Bone resorption releases calcium → confusion, weakness, lethargy, polyuria, constipation |
|---|---|---|
| R | Renal insufficiency | Bence Jones (free light chain) cast nephropathy, AL amyloid deposition, hypercalcemia-related damage; renal failure in up to 50% of patients |
| A | Anemia | Normocytic normochromic anemia from marrow replacement; leukopenia and thrombocytopenia may also occur |
| B | Bone lesions | Lytic "punched-out" defects 1-4 cm, most commonly in vertebral column, ribs, skull, pelvis, femur, clavicle, scapula; pathologic fractures are common |
Additional manifestations:
- Recurrent bacterial infections: Myeloma cells suppress normal B-cell function, severely depressing functional antibody production despite elevated total IgG (mostly non-functional M protein)
- Hyperviscosity syndrome: Particularly with IgA or IgM M proteins; causes visual changes, neurologic symptoms, bleeding
- Peripheral neuropathy: Especially with AL amyloidosis
- Rouleaux formation on blood smear with markedly elevated ESR
Bone Marrow Histology
The image below shows myeloma infiltrating the bone marrow - large, atypical plasma cells with prominent nucleoli, abnormal chromatin, and Russell bodies (cytoplasmic immunoglobulin inclusions):
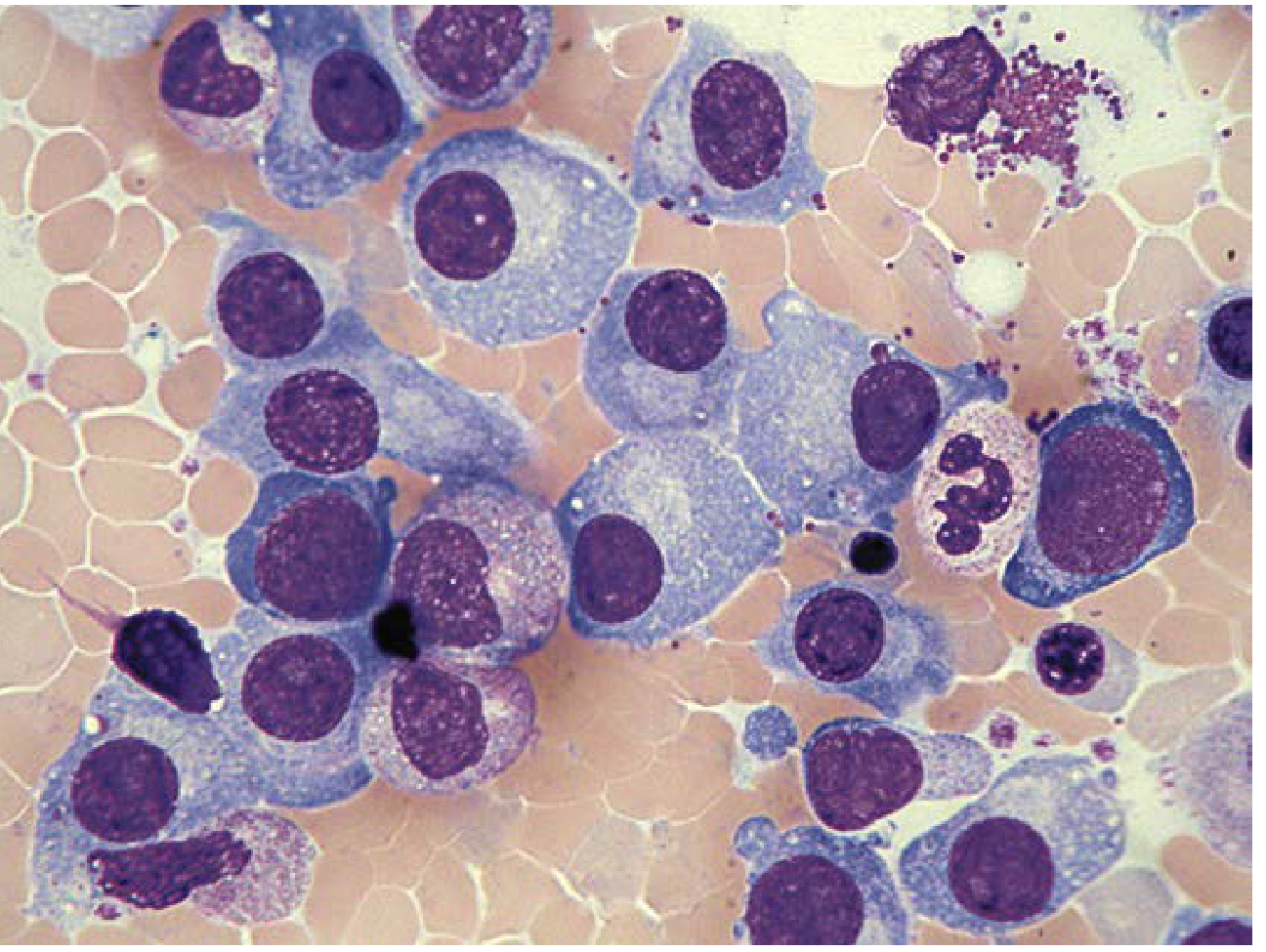
Bone marrow aspirate showing large neoplastic plasma cells - Henry's Clinical Diagnosis, p. 758
Morphologic findings:
- Plasma cells typically >30% of marrow cellularity in active disease
- Cells may show large nucleoli, cytoplasmic inclusions (Russell bodies), and varying degrees of anaplasia
- Immature (plasmablastic) variants carry worse prognosis
- In advanced disease, visceral spread and plasma cell leukemia can occur
Immunophenotype: CD38(+), CD138(+), loss of CD19 (normally present on plasma cells), CD20(-); monotypic light chain restriction
Myeloma Kidney
Renal involvement occurs through several mechanisms:
- Cast nephropathy (most important): Bence Jones proteins form obstructive casts in distal convoluted tubules and collecting ducts, surrounded by multinucleate giant cells; tubular epithelium adjacent to casts undergoes necrosis
- AL amyloidosis: Light chains deposit as amyloid in glomeruli and vessel walls
- Light chain deposition disease: Non-amyloid linear light chain deposits in glomeruli/interstitium
- Metastatic calcification: From hypercalcemia
- Bacterial pyelonephritis: From hypogammaglobulinemia
Renal failure is second only to infections as the leading cause of death in myeloma.
- Robbins & Kumar Basic Pathology, p. 422
Diagnostic Criteria
MGUS vs. Smoldering vs. Active Myeloma
| Feature | MGUS | Smoldering Myeloma | Active Myeloma |
|---|---|---|---|
| Serum M-protein | <30 g/L | ≥30 g/L | Any level |
| Marrow plasma cells | <10% | ≥10% | Clonal plasma cells present |
| CRAB features | None | None | Present |
| Treatment needed | No | Generally no | Yes |
Progression risk of smoldering myeloma:
- 10% per year for first 5 years
- 3% per year for years 5-10
- 1% per year thereafter
Definitive diagnosis requires:
- Bone marrow biopsy showing clonal plasma cells
- Serum/urine protein electrophoresis (SPEP/UPEP) with immunofixation to identify and type the M protein
- Serum free light chain assay
- Imaging (skeletal survey, CT, PET-CT or whole-body MRI) for bone lesions
- Henry's Clinical Diagnosis and Management, p. 758
Staging
Revised International Staging System (R-ISS):
| Stage | Criteria | Median OS |
|---|---|---|
| I | β2-microglobulin <3.5 mg/L + albumin ≥3.5 g/dL + standard cytogenetics + normal LDH | ~Not reached |
| II | Neither I nor III | ~83 months |
| III | β2-microglobulin ≥5.5 mg/L + high-risk cytogenetics [t(4;14), t(14;16), del(17p)] or elevated LDH | ~43 months |
Treatment
Newly Diagnosed - Transplant Eligible
Current standard of care uses triplet or quadruplet induction followed by autologous stem cell transplantation (ASCT):
- VRd: Bortezomib (proteasome inhibitor) + Lenalidomide (IMiD) + Dexamethasone
- Dara-VRd: Daratumumab (anti-CD38 monoclonal antibody) + VRd - increasingly adopted as preferred quadruplet regimen
- ASCT remains standard of care when feasible; early transplant vs. delayed transplant outcomes are comparable in overall survival but early transplant provides longer PFS
- Tandem ASCT may benefit high-risk patients
Newly Diagnosed - Transplant Ineligible
- VRd (reduced intensity) or DRd (Daratumumab + Lenalidomide + Dexamethasone)
- Continuous therapy until progression
Maintenance Post-ASCT
- Lenalidomide maintenance is standard; extends PFS and OS
- High-risk cytogenetics: add bortezomib
Relapsed/Refractory Multiple Myeloma (RRMM)
Key drug classes available:
- Proteasome inhibitors: Bortezomib, Carfilzomib, Ixazomib
- Immunomodulatory drugs (IMiDs): Thalidomide, Lenalidomide, Pomalidomide
- Anti-CD38 monoclonals: Daratumumab, Isatuximab
- Anti-SLAMF7: Elotuzumab
- Bcl-2 inhibitor: Venetoclax (t(11;14) myeloma)
- BCMA-targeted therapies (newer agents):
- CAR-T cells: Idecabtagene vicleucel (ide-cel), Ciltacabtagene autoleucel (cilta-cel)
- Bispecific antibodies: Teclistamab, Elranatamab, Talquetamab
A 2024 meta-analysis in J Immunother Cancer comparing CAR-T vs. bispecific antibodies as third-line or later treatment found comparable efficacy, informing sequencing decisions in RRMM.
Role of Allogeneic Transplantation
-
Limited role due to high treatment-related toxicity
-
Reduced-intensity allogeneic transplant may be considered in selected patients who relapse after ASCT
-
Goldman-Cecil Medicine, International Edition
Prognosis
- Without treatment, patients with multiple lytic lesions survive 6-12 months
- With modern therapy, median survival has improved to approximately 5-7+ years in some series
- Smoldering myeloma may require no treatment for many years
- Renal failure and infections are the leading causes of death
- High-risk cytogenetics [del(17p), t(4;14), t(14;16)] carry significantly worse prognosis
Clinical Variants
| Variant | Key Features |
|---|---|
| Smoldering (asymptomatic) myeloma | High M protein or marrow plasma cells but no CRAB; watch-and-wait |
| Nonsecretory myeloma | ~1% of cases; tumor cells do not produce/secrete Ig; diagnosis by marrow biopsy and FLC assay |
| Solitary plasmacytoma | Single bone or soft tissue lesion; treat with radiotherapy; monitor for progression to MM |
| Plasma cell leukemia | >20% circulating plasma cells; aggressive variant |
| POEMS syndrome | Polyneuropathy, Organomegaly, Endocrinopathy, M protein, Skin changes |
Summary
Multiple myeloma is a systemic plasma cell malignancy defined by the production of monoclonal immunoglobulin, bone marrow infiltration, and end-organ damage (CRAB). Treatment has been transformed by proteasome inhibitors, IMiDs, anti-CD38 antibodies, and most recently BCMA-directed CAR-T and bispecific antibody therapies. Recent PubMed evidence (2024-2026) focuses heavily on optimizing the role of CAR-T vs. bispecific antibodies in relapsed/refractory disease, with second primary malignancies after CAR-T and non-relapse mortality being important safety signals to monitor.
This is a shared conversation. Sign in to Orris to start your own chat.