Elaborate on the role of HLA B27, non HLA- like ERAP, aminopeptodases and IL123/IL17 axis in Spondyloarthritis. Detailed notes from rheumatology textbooks and journals
HLA-B27 ERAP1 spondyloarthritis pathogenesis
IL-23 IL-17 axis ankylosing spondylitis
PMID: 38024089
PMID: 38324125
Immunogenetics of Spondyloarthritis: HLA-B27, ERAP1/Aminopeptidases, and the IL-23/IL-17 Axis
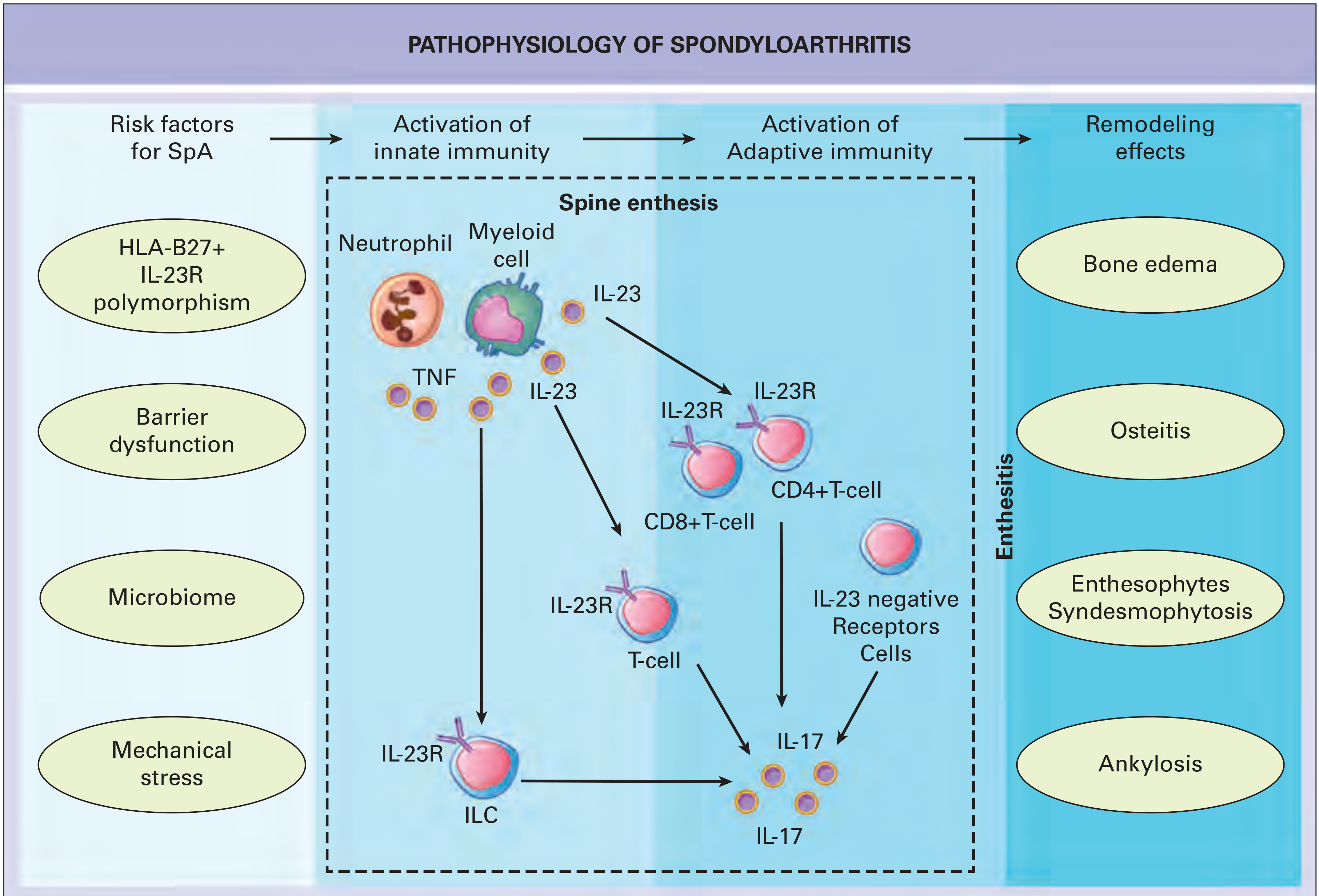
1. Overview: The Heritable Basis of SpA
- HLA-B27 (MHC Class I - dominant, ~20% of familial risk)
- Non-MHC aminopeptidases: ERAP1/ERAP2 (second strongest non-MHC signal)
- IL-23R and downstream IL-23/IL-17 pathway genes
2. HLA-B27: The Central Genetic Driver
2.1 Epidemiology and Association Strength
- ~80-95% of AS patients are HLA-B27+ (compared with 7% prevalence in Northern Europeans)
- Prevalence of HLA-B27 ranges from virtually absent in Australian Aboriginals, ~1% in Japan, ~7% in North America and Northern Europe, to ~50% in certain Western Canadian indigenous tribes
- Homozygous HLA-B27 carriers carry roughly doubled risk of AS vs. heterozygotes
- Despite this, the majority of HLA-B27 carriers never develop AS, indicating that HLA-B27 is necessary but not sufficient
2.2 HLA-B27 Subtypes: Which Are Arthritogenic?
| Subtype | Association with AS |
|---|---|
| B2705, B2704, B2702, B2703, B2707, B2708 | Positively associated |
| B*2706 (Southeast Asian) | Underrepresented/possibly protective |
| B*2709 (Sardinian) | Underrepresented/not protective |
2.3 The Four Mechanistic Hypotheses for HLA-B27 in SpA
A. Arthritogenic Peptide Hypothesis
B. Unfolded Protein Response (UPR) / HLA-B27 Misfolding Hypothesis
- XBP-1 (X-box binding protein-1)
- ATF-6 (activating transcription factor-6)
C. Free Heavy Chain (FHC) / Homodimer Hypothesis
- KIR3DL2 (killer cell immunoglobulin-like receptor) on NK cells and CD4+ T cells
- LILRB2 (leukocyte immunoglobulin-like receptor B2)
D. Innate Immune / Microbial Hypothesis
From Goldman-Cecil: "About 90% of patients with ankylosing spondylitis are B27-positive, so to a certain extent it may be necessary but not sufficient for the disease."
3. Non-HLA Genes: ERAP1, ERAP2, and the Aminopeptidase Family
3.1 ERAP1 - The Second Strongest Genetic Risk Factor
"The association with ankylosing spondylitis and ERAP1 is restricted to patients with HLA-B27+ ankylosing spondylitis, thereby suggesting a gene-gene interaction." - Goldman-Cecil Medicine
3.2 Biological Function of ERAP1
-
Peptide trimming for MHC Class I presentation: ERAP1 trims peptide precursors (8-16 amino acids) down to the optimal length (8-10 amino acids) for loading onto nascent MHC Class I molecules (including HLA-B27). The peptide repertoire presented on the cell surface is thus directly shaped by ERAP1 activity.
-
Control of the Unfolded Protein Response: ERAP1 also regulates ER homeostasis and the UPR - linking ERAP1 variants to HLA-B27 misfolding and enhanced IL-23 production.
3.3 How ERAP1 SNPs Drive Disease
- Loss-of-function ERAP1 variants generate longer, suboptimal peptides → altered self-peptide presentation on HLA-B27 → aberrant T cell responses
- Gain-of-function ERAP1 variants may over-trim peptides → reduced or altered presentation → immune dysregulation
- The net result: ERAP1 polymorphisms modify which peptides are loaded onto HLA-B27, shifting the self-peptide landscape toward arthritogenic sequences
3.4 ERAP2 and Other Aminopeptidases
From Firestein & Kelley's: "ERAP1 has also been associated with ERA [enthesitis-related arthritis]. Because ERAP1 encodes a protein product that controls the unfolded protein response and because HLA-B27 has been noted to be prone to misfolding, the combinations of these variants contribute to disease risk."
4. The IL-23/IL-17 Axis: The Cytokine Architecture of SpA
4.1 Genetics Supporting the IL-23 Pathway
| Gene | Locus | Function in SpA |
|---|---|---|
| IL23R | 1p31 | Acts on IL-23R-expressing T cells, ILCs, γδ T cells |
| IL6R | 1q21 | Th17 lymphocyte differentiation |
| RORC | 1q21 | Master transcription factor for Th17 differentiation |
| TYK2 | 19p13 | JAK kinase; IL-23 and IL-12 signaling |
| STAT3 | 17q21 | Signal transducer downstream of IL-23R and IL-6R |
| IL12B | 5q33 | p40 subunit shared by IL-12 and IL-23 |
From Rheumatology 2-Volume Set (Elsevier 2022): "In particular, genes in the interleukin-23 response pathway and genes encoding aminopeptidases involved in peptide processing before HLA class I presentation are overrepresented among ankylosing spondylitis-associated variants."
4.2 IL-23: The Upstream Master Regulator
- Myeloid cells (macrophages, dendritic cells)
- Intestinal epithelium and lamina propria macrophages
- Cells within the enthesis microenvironment
- CD4+ Th17 cells
- CD8+ Tc17 cells
- Innate lymphoid cells (ILCs) - type 3
- γδ T cells (particularly in entheseal tissue)
- Mucosal-associated invariant T (MAIT) cells
4.3 The Enthesitis-Based IL-23 Model
- Biomechanical stress at tendon insertions (entheses) initiates microtrauma
- Microtrauma activates innate immune cells at the enthesis, producing IL-23
- IL-23 acts on resident IL-23R+ cells in the enthesis - particularly CD4-/CD8- lineage-negative cells and later confirmed to be γδ T cells
- These cells produce IL-17A and IL-22 in response to IL-23 stimulation
- IL-17 drives osteoclast activation, periosteal new bone formation, and systemic inflammation
From Rheumatology 2-Volume Set: "The genetics of human SpA and its therapy points to a pivotal role for the IL-23-IL-17 axis. The IL-23-dependent enthesitis model as originally defined by Sherlock and colleagues described a CD4 and CD8 lineage negative cell population as being important in disease. More recently, another study in the same enthesitis model showed that gamma delta T cells that were IL-23 responsive were key players in disease pathogenesis."
4.4 IL-17 Family: Effector Cytokines in SpA
| Effect | Consequence in SpA |
|---|---|
| Neutrophil recruitment via CXCL1/CXCL8 induction | Innate amplification of inflammation |
| Synoviocyte and osteoblast activation | Joint inflammation and new bone formation |
| Periosteal osteoprogenitor stimulation | Enthesophyte and syndesmophyte formation |
| Induction of metalloproteases | Cartilage and bone erosion |
| IL-6 synergy | Th17 amplification loop |
| Stimulation of RANKL | Paradoxical osteoclast activation contributing to erosive disease |
4.5 ILC3s and γδ T Cells: The Innate IL-17 Source
- SpA inflammation is fundamentally an innate-predominant process (unlike RA)
- Anti-TNF therapy suppresses symptoms but does not prevent relapse on cessation because it does not address the underlying IL-23-driven innate activation
- Anti-IL-17 and anti-IL-23 therapies show efficacy in axSpA and PsA
4.6 The IL-23/IL-17 Bridge to Gut Microbiome and Barrier Dysfunction
- Prevotella copri and reduced Faecalibacterium prausnitzii characterize the AS gut microbiome
- Bacterial antigens translocating across a leaky gut mucosa stimulate macrophages to produce IL-23
- HLA-B27 itself alters gut microbiota composition in transgenic rat models (Brown MA 2018), suggesting a bidirectional interaction
4.7 IL-23/IL-17 and New Bone Formation
- Activation of periosteal mesenchymal stem cells and osteoprogenitors
- Endochondral ossification as the mechanism of syndesmophyte growth
- Enthesophyte formation at ligamentous insertions
5. Integration: How HLA-B27, ERAP, and IL-23/IL-17 Form One Unified Pathway
HLA-B27 misfolding (UPR)
↓
Enhanced ER stress → NF-κB activation → ↑IL-23 production
+
ERAP1 polymorphisms → altered peptide trimming → aberrant self-peptide presentation on HLA-B27
↓
Aberrant T cell (CD8+) and NK cell (via FHC) activation
+
Biomechanical stress + gut dysbiosis → innate IL-23 at enthesis/gut
↓
IL-23 acts on γδ T cells + ILCs at enthesis
↓
IL-17A / IL-17F / IL-22 production
↓
Inflammation (neutrophils, macrophages) + New bone formation (via Wnt/BMP)
↓
Enthesitis → Sacroiliitis → Syndesmophytosis → Ankylosis
6. Therapeutic Implications
| Target | Drug Class | Evidence in SpA |
|---|---|---|
| TNF | Anti-TNF (infliximab, adalimumab, etanercept) | Established for axSpA and PsA; suppresses but does not "cure" |
| IL-17A | Secukinumab, ixekizumab | Superior to TNF in some axSpA endpoints; highly effective in PsA |
| IL-17A/F | Bimekizumab | Emerging data in axSpA |
| IL-12/23 p40 | Ustekinumab | Effective in PsA; limited data in axSpA |
| IL-23 p19 | Risankizumab, guselkumab | Effective in PsA; clinical trials in axSpA underway |
| JAK (IL-23/IL-6 signaling) | Tofacitinib, upadacitinib, filgotinib | Approved in AS and PsA |
| ERAP1 | No clinical agent yet | Active therapeutic target under investigation |
From Rheumatology 2-Volume Set: "The question as to whether IL-23/17 axis blockers are superior to anti-TNF has not been resolved, and both remain first-line biologic choices depending on the dominant clinical phenotype."
7. Recent PubMed Evidence (2023-2026)
- Saad et al., Cureus 2023 (PMID 38024089): Comprehensive review of ERAP1 structure, SNPs, epistasis with HLA-B27, and association with axSpA clinical parameters. Positions ERAP1 as a potential therapeutic target.
- Srinath, Nakamura & Haroon, Curr Rheumatol Rep 2024 (PMID 38324125) - SPARTAN Meeting Proceedings: Type 3 immunity centered around IL-23/IL-17 is confirmed as a key driver of axSpA. HLA-B27 drives downstream T cell responses but alone is insufficient for disease. GWAS identifies immune activation pathways. Gut microbiome studies show altered flora migrating from gut to joint.
- Braun, Rudwaleit & Sieper, Z Rheumatol 2024 (PMID 38112753): 50-year retrospective on HLA-B27; reaffirms its diagnostic utility and reframes its mechanistic contributions.
- Bordbar et al., Best Pract Res Clin Rheumatol 2024 (PMID 39085016): Biology of HLA Class I-associated inflammatory diseases - cross-disease analysis including AS, Behcet's, and reactive arthritis.
- Parvin et al., Front Immunol 2025 (PMID 41019055): Inflammatory-osteogenic axis in AS; mechanisms of dysregulation and emerging therapeutic interventions with emphasis on IL-17/IL-23 and new bone formation.
Summary Table: Key Points
| Feature | Detail |
|---|---|
| Heritability of AS | >90% (twin studies) |
| HLA-B27 frequency in AS | 80-95% |
| Odds ratio for AS in B27 carriers | >50 (White Europeans) |
| Disease-associated subtypes | B2705, B2704, B2702, B2703 |
| Non-disease subtypes | B2706, B2709 |
| HLA-B27 mechanism 1 | Arthritogenic peptide (aberrant CTL activation) |
| HLA-B27 mechanism 2 | ER misfolding → UPR → NF-κB → IL-23 |
| HLA-B27 mechanism 3 | Free heavy chains → NK/KIR activation → IL-17 |
| ERAP1 locus | Chromosome 5q15 |
| ERAP1 epistasis | Only operates in HLA-B27+ AS |
| ERAP1 function | Trims peptides for MHC Class I loading; UPR regulation |
| IL23R locus | Chromosome 1p31 |
| Shared genetic overlap | AS, IBD, psoriasis (IL23R, IL12B, TYK2) |
| Primary IL-17 source at enthesis | γδ T cells + ILC3s (innate, not Th17) |
| IL-17 downstream effects | Neutrophil recruitment, new bone formation, osteoclast activation |
| New bone mechanism | Wnt/BMP activation, endochondral ossification |
| Anti-IL-17 drugs approved in SpA | Secukinumab, ixekizumab, bimekizumab |
Discuss in detail about CAR-T cells and their application in autoimmune diseases. From Kelley, hochberg and other textbooks and latest articles
CAR-T cell therapy autoimmune disease
CAR-T cell systemic lupus erythematosus clinical trial
CAR-T cell therapy autoimmune diseases 2024 2025 clinical trials results rheumatology
PMID: 37748491
PMID: 39107407
PMID: 38777376
PMID: 40446794
PMID: 40993243
PMID: 41028170
PMID: 37495877
PMID: 40681435
CAR-T cell therapy myasthenia gravis multiple sclerosis neuromyelitis optica
CAR-Treg regulatory T cell therapy rheumatoid arthritis autoimmune REGULATE-RA 2025
CAR-T Cells and Their Application in Autoimmune Diseases
1. CAR-T Cell Biology: Structure and Mechanism
1.1 What is a CAR?
- The antigen-binding domain - typically a single-chain variable fragment (scFv) derived from the heavy (VH) and light (VL) chains of a monoclonal antibody
- A hinge/spacer domain - provides flexibility
- A transmembrane domain - anchors the receptor to the T cell membrane
- An intracellular signaling domain - mimics both TCR activation and co-stimulation
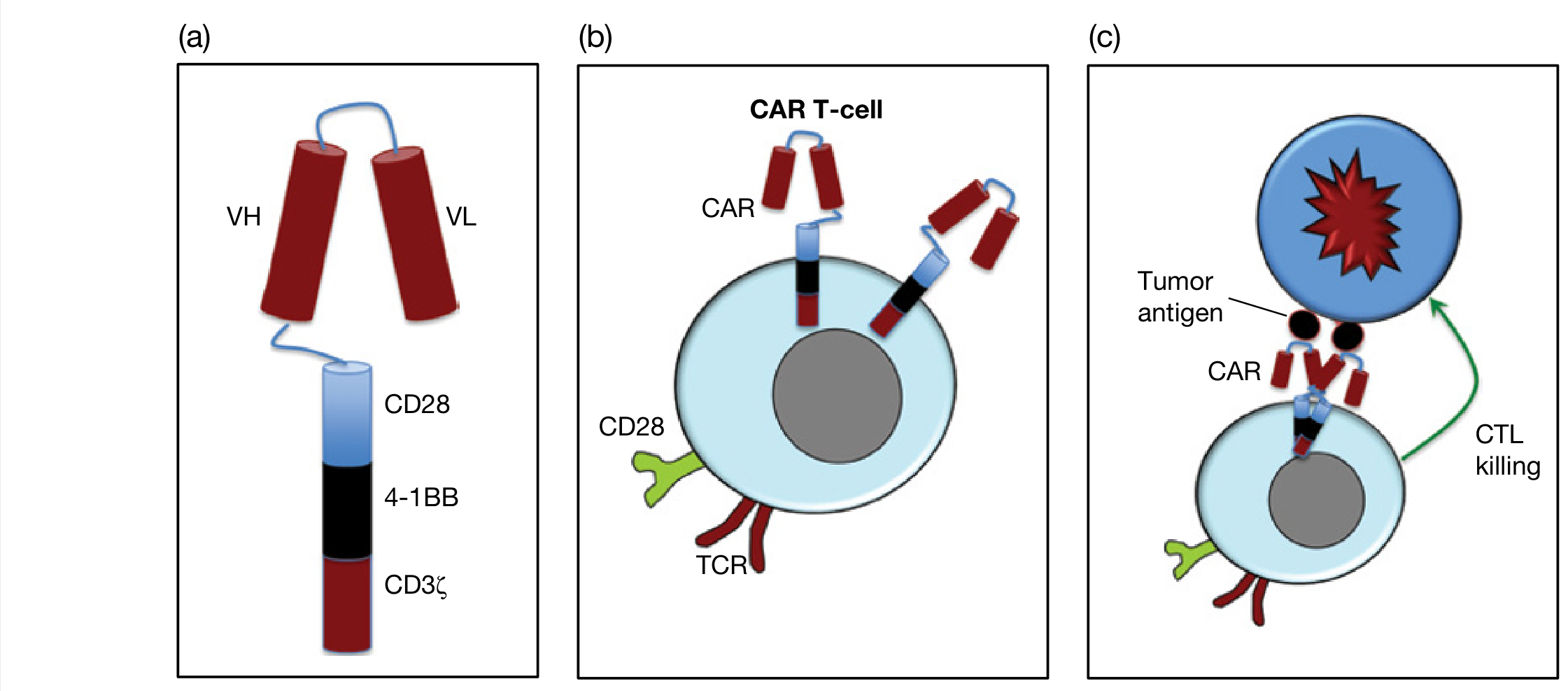
From Roitt's Essential Immunology: "CARs are constructed by combining the antigen-binding site of a monoclonal antibody directed against a tumor antigen, with some of the intracellular signaling components of a T-cell receptor (e.g., CD3ζ chain). This artificial receptor enables antigen recognition on the tumor independent of MHC restriction, while retaining the desirable antitumor properties of a T-cell."
1.2 Generations of CAR Design
| Generation | Signaling Components | Key Feature |
|---|---|---|
| 1st | CD3ζ only | Limited in vivo persistence; poor efficacy |
| 2nd | CD3ζ + CD28 or 4-1BB | Current clinical standard; improved persistence |
| 3rd | CD3ζ + CD28 + 4-1BB or OX40 | Enhanced signaling; under investigation |
| 4th ("armored" CAR) | 3rd gen + cytokine payload (e.g. IL-15) | Self-sustaining; overcomes immunosuppressive microenvironment |
| 5th | Incorporates JAK-STAT signaling domains | Most potent; T cell memory induction |
From Roitt's Essential Immunology: "To work, the CAR intracytoplasmic domain needs to mimic both natural TCR stimulation, as well as co-stimulation via CD28. This is achieved through combining components from the CD28 co-receptor as well as the immunoreceptor tyrosine-based activation motifs (ITAMs) present in the cytoplasmic CD3ζ domain."
1.3 Manufacturing: Autologous vs. Allogeneic CAR-T
- Patient T cells collected by leukapheresis
- Activated ex vivo
- Transduced with CAR-encoding vector (retroviral or lentiviral)
- Expanded in culture over 10-14 days
- Quality-tested and infused after lymphodepletion conditioning (typically fludarabine + cyclophosphamide)
2. The Rationale for CAR-T in Autoimmune Disease
2.1 Why B Cells Are the Target
- They produce pathogenic autoantibodies (anti-dsDNA in SLE, anti-AChR in myasthenia gravis, anti-aquaporin-4 in NMOSD, ACPA in RA)
- They present autoantigens to autoreactive T cells
- They secrete pro-inflammatory cytokines (IL-6, IL-12, TNF, lymphotoxin)
- Long-lived plasma cells (LLPCs) in bone marrow survive conventional B cell-depleting therapies and maintain autoantibody production for years
From the Lancet (Schett et al. 2023, PMID 37748491): "B-cell-depleting monoclonal antibodies, such as rituximab, have poor therapeutic efficacy in autoimmune diseases, mainly due to the persistence of autoreactive B cells in lymphatic organs and inflamed tissues."
2.2 The Concept of "Immune Reset"
- Active trafficking to lymphoid organs, bone marrow niches, and inflamed tissue - sites that rituximab cannot penetrate effectively
- In vivo expansion - each CAR-T cell can proliferate 1,000-fold after antigen engagement
- Long-term persistence - memory CAR-T cells can surveil for months to years
- Complete elimination of LLPCs (via BCMA-targeting), eliminating the autoantibody "memory reservoir"
From Nat Rev Rheumatol (Schett et al. 2024, PMID 39107407): "The scientific rationale behind this approach is that deep depletion of B cells, including autoreactive B cell clones, could restore normal immune function, referred to as an immune reset."
2.3 Targets in Autoimmune CAR-T Therapy
| Target | Cells Eliminated | Diseases Targeted |
|---|---|---|
| CD19 | All B cell lineage (naive, memory, some plasma cells) | SLE, SSc, IIM, MS, NMOSD, MG, RA |
| BCMA (CD269) | Plasmablasts and long-lived plasma cells | SLE (especially with LN), IIM, SSc |
| CD19 + BCMA dual | B cells + LLPCs simultaneously | Refractory SLE, SLE with LN |
| CD20 | B cells (overlapping with CD19) | B cell malignancies; being explored in AD |
3. Clinical Evidence by Disease
3.1 Systemic Lupus Erythematosus (SLE) - The Pivotal Disease
- First reported 2022 in Nature Medicine (Mackensen A et al.): 5 patients with refractory SLE, all achieved drug-free remission (DORIS criteria)
- Follow-up in N Engl J Med 390:687, 2024 (case series): Sustained remission confirmed with complete serological normalization (anti-dsDNA clearance, complement normalization)
- The CASTLE basket study (NCT06347718) is ongoing, recruiting refractory SLE, SSc, and IIM
- 12 patients with SLE/lupus nephritis (LN classes III-V)
- Endpoint: simultaneous reset of humoral and B cell immunity
- Results: SLEDAI-2K reduced from 10.6 to 2.7 at 3 months; all patients negative for autoantibodies including LLPC-derived antibodies; medication-free remission achieved in all patients with follow-up up to 46 months
- Only mild cytokine release syndrome (CRS)
- 15 patients (14F, 1M) with treatment-refractory SLE
- Fludarabine/cyclophosphamide lymphodepletion followed by co-infusion of autologous anti-CD19 + anti-BCMA CAR-T cells
- Results at week 12: 80% (12/15) fulfilled both LLDAS and DORIS remission criteria
- Grade 1 CRS in 86.7%; no neurotoxicity, no treatment-related deaths
- Multiomic analyses: complete elimination of autoreactive CD19+BCMA+ clones; reconstitution of naive IgM/IgD B cells; durable downregulation of IFN-stimulated and BAFF-dependent signatures
- Median follow-up 712 days; 3 patients monitored 1 year showed "sustained eradication of pathogenic clones, suggesting potential cure"
- 3 patients with severe refractory SLE with multi-organ involvement
- Off-the-shelf healthy-donor-derived anti-CD19 CAR-T cells
- No GvHD, no CRS, no ICANS; CAR-T expanded in vivo, peaking day 14
- SRI-4 remission at last visit in all patients; SLEDAI scores declined significantly
- Establishes feasibility of allogeneic strategy, removing manufacturing barriers
- 7 biopsy-confirmed LN patients; median follow-up 9 months
- SLEDAI-2K: median 18 at baseline → 0 at last follow-up
- 5/7 patients achieved DORIS complete remission
- Renal biopsy showed decreased immune complex deposition; transcriptomics showed reduced clonal abundance and proinflammatory status
- Confirms BCMA-only strategy is viable (not always necessary to combine with CD19)
- Preliminary results of CTA313 in active SLE: encouraging safety, efficacy, and cellular kinetics; by targeting both B cells and plasma cells simultaneously, potential new option for refractory lupus.
3.2 Systemic Sclerosis (SSc/Scleroderma)
From Harrison's Principles of Internal Medicine 22E (2025): "The rapid clinical response, fibrosis resolution, and vascular regeneration observed in some SSc patients treated with immunomodulatory or immunoablative and chimeric antigen receptor (CAR) T-cell (CAR-T) therapies."
- Auth J et al., Lancet Rheumatol 2025;7:83-93: CD19-targeting CAR-T in diffuse SSc case series - skin fibrosis improvement (mRSS decline), ILD stabilization
- CASTLE study includes SSc as one of its basket arms
3.3 Idiopathic Inflammatory Myopathies (IIM) - Dermatomyositis/Polymyositis
- Anti-CD19 CAR-T induced complete clinical and serological remission in refractory dermatomyositis
- Myositis-specific antibodies (anti-MDA5, anti-Mi-2) cleared post-treatment
- Muscle enzymes normalized
- The CASTLE study includes IIM
3.4 Neuromyelitis Optica Spectrum Disorder (NMOSD) and Multiple Sclerosis (MS)
- Phase 1 data (Haghikia A, Schett G, Mougiakakos D, Lancet Neurol 2024, PMID 38760099): Anti-CD19 CAR-T in neuroimmunological diseases including NMOSD and MS. First patients showed B cell depletion, clinical stabilization, and reduced relapse rates.
- Harrison's 2025 includes a further reading reference: "Single-cell analysis of anti-BCMA CAR T cell therapy in patients with central nervous system autoimmunity. Sci Immunol"
- JAMA Neurol 2025 (Ismail et al., PMID 39585688) reviewed current and future roles in neurology
3.5 Myasthenia Gravis (MG)
- Preclinical and early clinical data support CD19 and BCMA-directed CAR-T
- Included in the Nat Rev Rheumatol 2024 Schett review as a candidate condition with early data
3.6 Rheumatoid Arthritis (RA) - CD19 CAR-T and the Novel CAR-Treg Approach
- KYV-101 is a fully human autologous CD19 CAR-T with CD28 co-stimulation
- COMPARE trial Phase 1/2 (Charite, Berlin): robust CAR-T expansion and B-cell depletion, well-tolerated profile, compelling outcomes in difficult-to-treat RA
- Phase 2 randomized portion enrolled; enrollment completed as of ACR 2025
- Freeley M, Clin Rev Allergy Immunol 2025 (PMID 41258618) provides a comprehensive review of CAR-T in RA
- SBT-77-7101 (Sonoma Biotherapeutics): Autologous Tregs transduced with a CAR targeting citrullinated peptide (CitP) - the autoantigen driving RA
- Mechanism: CitP-specific CAR Tregs home to joints where citrullinated proteins are expressed, locally suppress inflammation without global immunosuppression
- No lymphodepletion required - unlike conventional CAR-T, CAR-Treg cells are not cytotoxic
- Phase 1 results (ACR 2025): 6 patients, infusions well-tolerated, no CRS, no neurotoxicity, no dose-limiting toxicities; substantial reductions in swollen joint counts and disease activity scores within 4 weeks; deeper responses at higher doses; anti-inflammatory effects in synovial tissue biopsies
- Phase 2 enrollment ongoing
4. Types of CAR Strategies in Autoimmune Disease
4.1 Conventional CAR-T (Cytotoxic) - B Cell Depletion
4.2 CAR-Treg - Localized Immune Suppression
- REGULATE-RA (CitP-CAR Tregs in RA) - Sonoma Biotherapeutics
- HLA-A2 CAR Tregs for kidney transplant rejection - Sangamo (NCT04817774)
- Liver transplant CAR Tregs - Quell Therapeutics (NCT05234190)
4.3 In Vivo CAR-T: The Next Frontier
From Nat Rev Drug Discov 2026 (Bot et al., PMID 41028170): "In vivo CAR-T cell engineering, in which CAR-T cells are generated directly inside the patient's body, seeks to overcome these challenges by eliminating the need for ex vivo cell processing... Recent advances in virology, RNA medicines and nanotechnology have catalysed a radical overhaul of this approach, which uses targeted delivery systems such as lentiviral vectors and lipid nanoparticles to introduce CAR-encoding genetic material into endogenous T cells."
5. Lymphodepletion Conditioning: Why It Is Needed
- Creates space in lymphoid niches for CAR-T expansion
- Removes regulatory cells and homeostatic cytokine sinks (IL-7, IL-15 increase after lymphodepletion, driving CAR-T proliferation)
- Reduces the autoreactive immune environment that the CAR-T must overcome
- Enhances in vivo expansion of the infused CAR-T product
6. Adverse Effects and Safety Profile
6.1 Cytokine Release Syndrome (CRS)
- In autoimmune diseases, CRS tends to be milder than in hematologic malignancies (lower tumor burden; less antigen load)
- Typically Grade 1-2 in autoimmune trials; Grade 3-4 rare
- Management: tocilizumab (anti-IL-6R), corticosteroids
- BCMA-CD19 compound CAR-T (PMID 38777376): "cCAR T therapy was well tolerated with mild cytokine release syndrome"
- Dual CD19+BCMA (Nat Med 2025): Grade 1 CRS in 86.7%; no grade 3+ CRS, no neurotoxicity, no deaths
6.2 ICANS (Immune Effector Cell-Associated Neurotoxicity Syndrome)
- Rare in autoimmune disease trials reported to date
- No ICANS observed in the allogeneic SLE trial (PMID 40446794) or BCMA-LN trial (PMID 40681435)
6.3 B Cell Aplasia and Hypogammaglobulinemia
- Transient hypogammaglobulinemia managed with IVIG replacement
- Long-term: healthy naive B cell reconstitution without return of autoreactive clones = the therapeutic goal
From Harrison's Principles (2025): "Anti-CD20 monoclonal antibodies, CAR-T therapy, bispecific T-cell engagers, and ICIs have all been associated with long-term B-cell aplasia that often..."
6.4 Cytopenias from Lymphodepletion
- Neutropenia (Grade 3-4: 100% in some trials, reversible)
- Thrombocytopenia (40%)
- Anemia (13-20%)
- All reversible with supportive care (G-CSF, transfusions)
6.5 Infection Risk
6.6 Graft-vs-Host Disease (GvHD) - Allogeneic Only
7. Comparison with Conventional B Cell-Directed Therapies
| Feature | Rituximab (anti-CD20) | CAR-T (anti-CD19) | CAR-T (anti-BCMA) |
|---|---|---|---|
| Mechanism | Ab-mediated ADCC, CDC | Direct CTL killing | Direct CTL killing |
| Plasma cell elimination | Poor | Partial (CD19+ plasma cells) | Complete (LLPCs) |
| Tissue penetration | Poor | Active trafficking | Active trafficking |
| In vivo expansion | None | 1000-fold expansion | 1000-fold expansion |
| Duration of effect | Months; repeat dosing needed | Potentially years | Potentially years |
| Autoantibody clearance | Incomplete | Complete in trials | Complete in trials |
| Immune reset | No | Yes (naive B cell reconstitution) | Yes + autoantibody memory cleared |
| CRS risk | Mild | Mild-moderate (manageable) | Mild |
| Cost/complexity | Low | High (currently) | High (currently) |
8. The Broader Vision: CAR-T as a "Living Drug" and One-Shot Therapy
From Nature 2023 (Baker DJ, June CH et al., PMID 37495877): "Engineering a patient's own T cells to selectively target and eliminate tumour cells has cured patients with untreatable haematologic cancers. Evidence from clinical and preclinical studies underscores the potential of CAR T therapy beyond oncology in treating autoimmunity, chronic infections, cardiac fibrosis, senescence-associated disease and other conditions."
From Roitt's Essential Immunology: "Additionally, these therapies have the potential to be 'one-shot' treatments that induce complete and sustained remission."
From Harrison's Principles of Internal Medicine 22E (2025): "Continued expansion of in vivo genome editing; and application of CAR-T technology to solid tumors and autoimmune disorders (e.g., systemic lupus erythematosus)."
9. Current Landscape and Future Directions
9.1 Clinical Trial Landscape (as of May 2025)
- 56 clinical trials registered for CAR-T in autoimmune rheumatic diseases (ARDs) as of May 2025 (Frontiers 2025 analysis)
- Phase I trials dominate (64%), confirming early-stage exploration
- 25 new trials registered in 2024 alone (peak year)
- Disease areas: SLE (most), SSc, IIM, Sjögren's syndrome, RA, ANCA vasculitis, myositis, antisynthetase syndrome
9.2 Next Generation Approaches
- Dual-targeting (CD19+BCMA simultaneously): eliminates both B cells and LLPCs; multiple trials ongoing
- Allogeneic "off-the-shelf" CAR-T: removes manufacturing delays, reduces cost; early safety data encouraging
- In vivo CAR-T (lipid nanoparticles/lentiviral vectors): eliminates need for leukapheresis; game-changing for accessibility
- CAR-Treg: antigen-specific suppression instead of depletion; no lymphodepletion; REGULATE-RA as proof-of-concept
- Armored/4th generation CAR-T: IL-15 or IL-21 co-expression improves persistence and efficacy
- Tolerance-inducing CAR-T: expressing FOXP3 or anti-inflammatory payloads at the lesion site
9.3 Unresolved Challenges
- Long-term safety data (follow-up mostly <4 years)
- Optimal patient selection (when to use in disease course)
- Durability of immune reset: will autoreactive clones re-emerge?
- Manufacturing complexity, cost, and access (especially in low/middle-income countries)
- The role of lymphodepletion conditioning in a vulnerable immunosuppressed population
- Regulatory frameworks for living drugs in non-oncology indications
Summary Table: CAR-T in Autoimmune Diseases at a Glance
| Disease | Main Target | Stage | Key Outcome |
|---|---|---|---|
| SLE | CD19, BCMA, CD19+BCMA | Phase 1; multiple trials | Drug-free DORIS remission in >80% in best trials; autoantibody clearance |
| Lupus Nephritis | BCMA, CD19+BCMA | Phase 1 | SLEDAI 18→0; DORIS complete remission in 5/7; renal biopsy improvement |
| SSc | CD19 | Case series/Phase 1 | Fibrosis resolution, vascular regeneration (Harrison's 2025) |
| IIM/Dermatomyositis | CD19 | Phase 1 (CASTLE) | Complete clinical + serological remission |
| MS/NMOSD | CD19, BCMA | Phase 1 | B cell depletion, clinical stabilization, reduced relapses |
| MG | CD19, BCMA | Early/preclinical | Autoantibody source elimination |
| RA (cytotoxic) | CD19 (KYV-101) | Phase 1/2 | B cell depletion, disease activity reduction (COMPARE trial) |
| RA (CAR-Treg) | CitP-CAR Treg | Phase 1 | Safe, no CRS, swollen joint reduction, synovial anti-inflammatory effects (REGULATE-RA) |
- Schett G et al., Lancet 2023;402:2031-40 (PMID 37748491)
- Schett G, Müller F et al., Nat Rev Rheumatol 2024;20:579-591 (PMID 39107407)
- Feng J et al., Nat Med 2025 (PMID 40993243) - Dual CD19+BCMA Phase 1
- Wang W et al., Ann Rheum Dis 2024 (PMID 38777376) - BCMA-CD19 cCAR Phase 1
- Wang D et al., Med 2025 (PMID 40446794) - Allogeneic CD19 CAR-T
- Hu Z et al., Ann Rheum Dis 2025 (PMID 40681435) - BCMA-only LN
- Baker DJ, June CH et al., Nature 2023 (PMID 37495877) - CAR-T beyond cancer
- Bot A et al., Nat Rev Drug Discov 2026 (PMID 41028170) - In vivo CAR-T
- REGULATE-RA (SBT-77-7101 CAR-Treg), Late-Breaking ACR 2025
- Harrison's Principles of Internal Medicine 22E (2025), Ch. 372 (SSc); Ch. 54 (gene/cell therapy)
- Roitt's Essential Immunology, Chapter 16 (Adoptive Immunotherapy)
- Firestein & Kelley's Textbook of Rheumatology (Ref. 127: Mackensen A et al., Nat Med 2022)