Wilson disease
Reading File
Finding Sources
Searching PubMed
Wilson disease diagnosis treatment
Reading File
Reading File
Reading File
Loading Image
Searching Images
Kayser-Fleischer ring Wilson disease cornea
Finding Sources
Reading File
Loading Image
I now have comprehensive information to provide a thorough answer on Wilson disease. Let me synthesize it.
Wilson Disease (Hepatolenticular Degeneration)
Overview
Wilson disease (WD) is an autosomal recessive disorder caused by loss-of-function mutations of the ATP7B gene on chromosome 13, which encodes a copper-transporting transmembrane ATPase. The mutation results in:
- Failure to excrete copper into bile (primary excretory route)
- Failure to incorporate copper into apoceruloplasmin → low serum ceruloplasmin
Prevalence: ~1 in 30,000; carrier frequency ~1 in 90. Symptoms typically appear between 6 and 40 years of age.
Pathophysiology
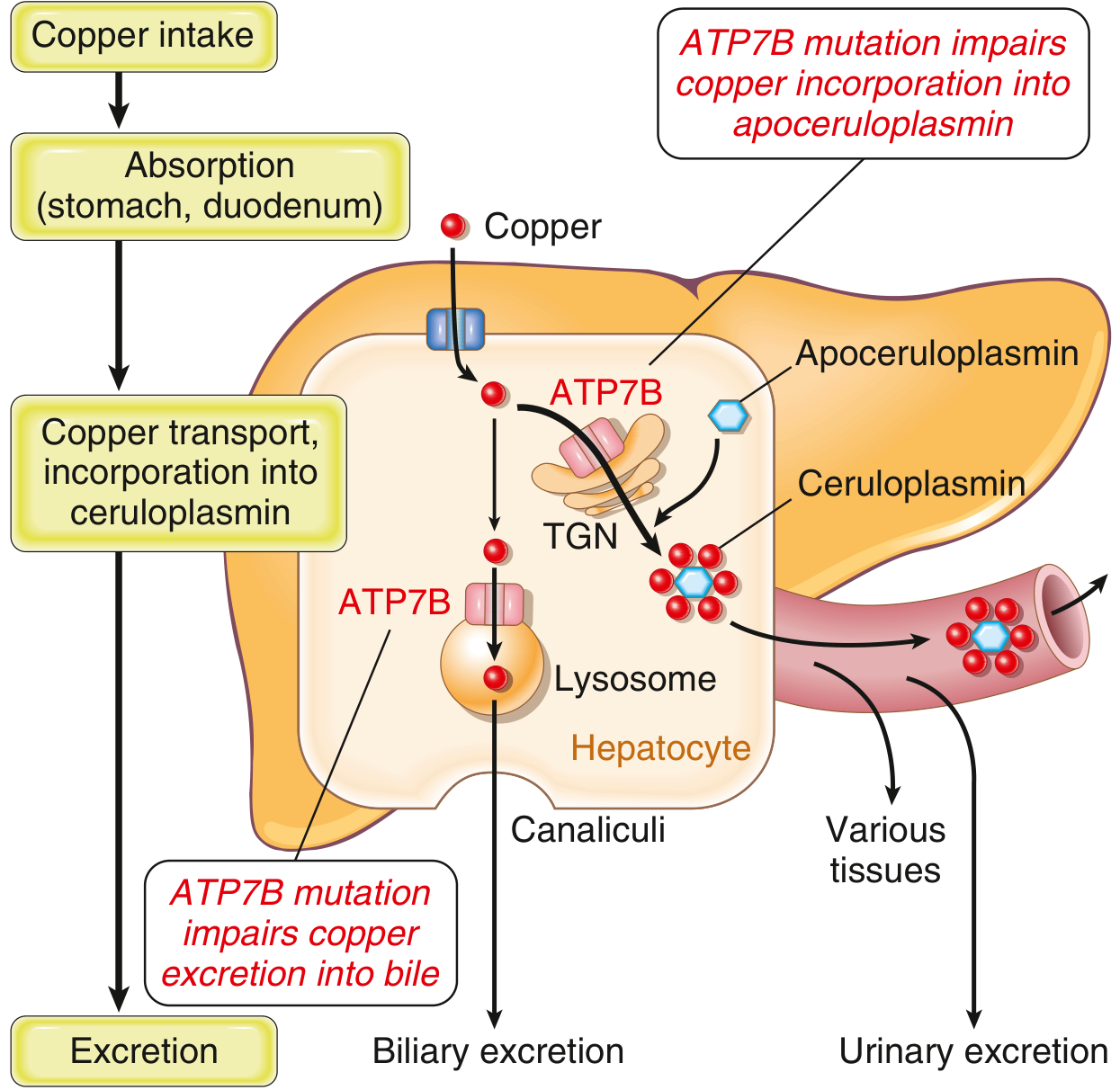
Normally, 40–60% of ingested copper (2–5 mg/day) is absorbed in the duodenum and transported to the liver complexed with albumin and histidine. In hepatocytes, ATP7B mediates two functions:
- In the trans-Golgi network: loads copper into apoceruloplasmin → ceruloplasmin secreted into blood
- In lysosomes: transports non-ceruloplasmin copper to bile canaliculi for biliary excretion
In Wilson disease, both pathways fail → copper accumulates in hepatocyte cytoplasm and lysosomes → increased ROS → hepatocyte injury. As copper overflows, non-ceruloplasmin-bound copper is released into the circulation, causing:
- Red cell hemolysis
- Copper deposition in brain (especially basal ganglia), corneas, kidneys, bones, joints, and parathyroid glands
- Elevated urinary copper excretion
— Robbins & Kumar Basic Pathology, p. 2018
Clinical Presentation
Patients present in one of three ways (roughly equally distributed):
| Presentation | Features |
|---|---|
| Hepatic | Steatosis, acute hepatitis, chronic hepatitis, fulminant hepatic failure (ALF), cirrhosis |
| Neurological | Tremor (wing-beating), chorea, dystonia, dysarthria, dysphagia, ataxia, gait disturbance, sardonic smile |
| Psychiatric | Depression (~30%), personality/mood changes, bipolar spectrum (~20%), psychosis, suicidal ideation (5–15%) |
About 70% of WD patients develop psychiatric symptoms over the long-term disease course, even with treatment. The cognitive profile is consistent with frontosubcortical network disturbance.
Key Signs
Kayser-Fleischer (KF) rings — pathognomonic ocular sign:
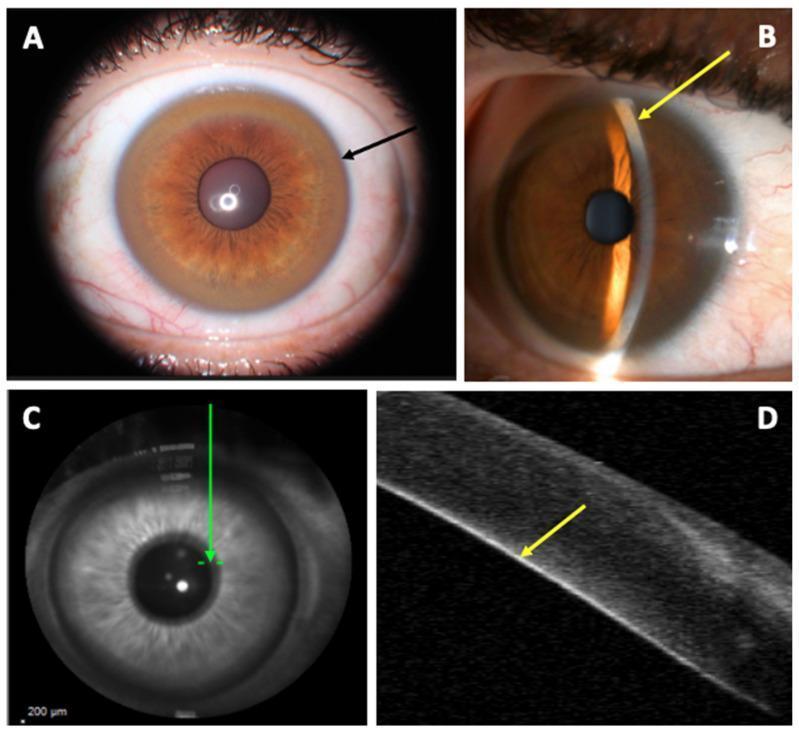
- Yellow-brown copper deposits in Descemet's membrane at the corneal limbus
- Present in 98% of patients with neurological WD; ~80% of all WD cases
- Best visualized by slit-lamp examination
- Can be absent in purely hepatic presentations
Sunflower cataract — another ocular finding (less specific)
Acute Liver Failure in Wilson Disease
A distinctive ALF presentation typically occurs in the second decade. Diagnostic clues:
- Coombs-negative hemolytic anemia (non-immune hemolysis)
- Serum alkaline phosphatase/total bilirubin < 4
- Serum AST/ALT > 2.2
- Unconjugated hyperbilirubinemia
— Sleisenger & Fordtran's GI and Liver Disease
Histopathology (Liver)
Changes range from mild to severe:
- Early: steatosis, focal hepatocyte necrosis
- Active disease: chronic hepatitis with moderate-severe inflammation, hepatocyte ballooning, Mallory hyaline bodies, steatohepatitis-like picture
- Late: cirrhosis
- Copper deposition demonstrable with special stains (rhodanine, orcein)
Toxic injury in the brain primarily affects the basal ganglia (putamen, caudate).
Diagnosis
No single test is reliably diagnostic; a combination is required:
| Test | Finding in Wilson Disease |
|---|---|
| Serum ceruloplasmin | Low (<20 mg/dL); hallmark but not diagnostic alone |
| 24-hour urine copper | Elevated (>100 µg/day symptomatic; >40 µg/day in children) |
| Slit-lamp exam | Kayser-Fleischer rings |
| Liver biopsy + hepatic copper | >250 µg/g dry weight |
| Serum "free" (non-ceruloplasmin) copper | Elevated |
| ATP7B mutation analysis | Confirms diagnosis; important for family screening |
Leipzig scoring system (combines clinical, biochemical, and genetic criteria) — scores ≥4 indicate Wilson disease.
Brain MRI: abnormal T2 signal in putamen, midbrain, pons, thalamus, cerebellum; atrophy common with neurological involvement.
Genetic testing is important for:
- Distinguishing carriers (heterozygotes) from affected homozygotes
- Screening first-degree relatives
- Populations with limited mutation repertoires (e.g., Sardinia — prevalence 1 in 7,000)
Treatment
Three established treatments; therapy must be lifelong:
1. D-Penicillamine
- Copper chelator; promotes urinary excretion
- 20–50% of neurological WD patients experience acute neurological worsening after initiation — a significant limitation
- Some patients do not recover to pre-treatment baseline
2. Trientine (Triethylene tetramine)
- Copper chelator
- Preferred over penicillamine due to lower rate of neurological worsening and fewer side effects
- Now more widely used as first-line
3. Zinc Salts (acetate, sulfate, gluconate)
- Mechanism: induces intestinal metallothionein → blocks copper absorption
- Not a chelator; used for presymptomatic patients and maintenance therapy
- Can be combined with chelation
- A phase III trial of zinc salt showed neurological improvement with fewer deteriorations vs. historical zinc/penicillamine/trientine data
4. Tetrathiomolybdate (ammonium)
- Blocks copper absorption and binds circulating copper
- Experimental but nearing clinical application
- May cause less neurological worsening than penicillamine
Liver Transplantation
- Indicated for ALF refractory to medical therapy or end-stage cirrhosis
- Corrects the metabolic defect (the transplanted liver has normal ATP7B)
Special situations
- Pregnancy: Treatment must continue throughout; postpartum hepatic decompensation may occur if stopped. Many successful pregnancies documented on D-penicillamine.
- Presymptomatic patients (identified via family screening): zinc or low-dose chelation
— Sleisenger & Fordtran's GI and Liver Disease; Bradley & Daroff's Neurology in Clinical Practice; Goodman & Gilman's Pharmacological Basis of Therapeutics
Prognosis
- Early treatment can lead to partial/full reversal of hepatic and neurological changes, including improvement of MRI abnormalities
- Without treatment, the disease is progressive and fatal
- Presymptomatic treatment (via family screening) prevents clinical manifestations entirely
This is a shared conversation. Sign in to Orris to start your own chat.