Myocardial infraction from pathology by harsh mohan
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Loading Image
Loading Image
I now have all the content needed. Let me compile a comprehensive response covering everything in Harsh Mohan's style (which follows Robbins closely).
Myocardial Infarction - Pathology (Robbins/Harsh Mohan Style)
Note: Harsh Mohan's Textbook of Pathology is not in this library's catalog. The content below is drawn from Robbins & Cotran Pathologic Basis of Disease and Robbins Basic Pathology - the same primary sources Harsh Mohan's textbook is based on, so the content is equivalent for exam purposes.
Definition
Myocardial infarction (MI), commonly called a "heart attack," is necrosis (death) of cardiac muscle resulting from prolonged ischemia. The 2018 joint cardiology task force defines it as "acute myocardial injury detected by abnormal cardiac biomarkers in the setting of evidence of acute myocardial ischemia."
Epidemiology
- ~800,000 MIs per year in the USA (nearly one every 40 seconds)
- ~400,000 deaths annually
- 10% occur before age 40; 45% before age 65
- Males at greater risk in middle age; gap narrows post-menopause
- Post-menopausal estrogen decline worsens coronary artery disease - IHD is the leading cause of death in older women
Etiology & Pathogenesis
Coronary Arterial Occlusion (90% of cases)
The following sequence underlies most MIs:
- An atheromatous plaque is disrupted or eroded - by endothelial injury, intraplaque hemorrhage, or mechanical forces - exposing subendothelial collagen and necrotic plaque contents to blood
- Platelets adhere, aggregate, and are activated, releasing thromboxane A2, ADP, and serotonin - causing further platelet aggregation and vasospasm
- Coagulation is activated by exposure of tissue factor, adding to the growing thrombus
- Within minutes, the thrombus completely occludes the coronary artery lumen
Angiography within 4 hours of MI onset shows thrombosis in ~90% of cases. At 12-24 hours without intervention, only 60% show thrombosis - meaning some occlusions clear spontaneously. This gives the rationale for early thrombolysis/angioplasty to limit necrosis.
Non-Atherosclerotic MI (~10% of cases)
- Vasospasm (with or without atherosclerosis) - cocaine, ephedrine
- Embolism - from left atrial mural thrombus (in AF), valve vegetations, prosthetic material, or paradoxical emboli via patent foramen ovale
- Vasculitis, amyloid deposition, sickle cell disease (intramyocardial arteriole disease)
Myocardial Response to Ischemia
Temporal Sequence
| Event | Time |
|---|---|
| Onset of ATP depletion | Seconds |
| Loss of contractility | < 2 minutes |
| ATP reduced to 50% of normal | 10 minutes |
| ATP reduced to 10% of normal | 40 minutes |
| Irreversible cell injury (necrosis) | 20-40 minutes |
| Microvascular injury | > 1 hour |
| Complete infarction | 6-12 hours |
- Aerobic metabolism ceases within seconds → drop in ATP, accumulation of lactic acid
- Contractility is lost within 1-2 minutes (before cell death)
- Irreversible damage occurs after 20-40 minutes of severe ischemia (flow ≤10% of normal)
- Subendocardial zone is the most vulnerable - it is the last to receive blood from epicardial vessels, and exposed to the highest intramural pressure
- A narrow rim (~0.1 mm) of subendocardium directly beneath the lumen is spared by diffusion from the ventricular cavity
Patterns of Infarction
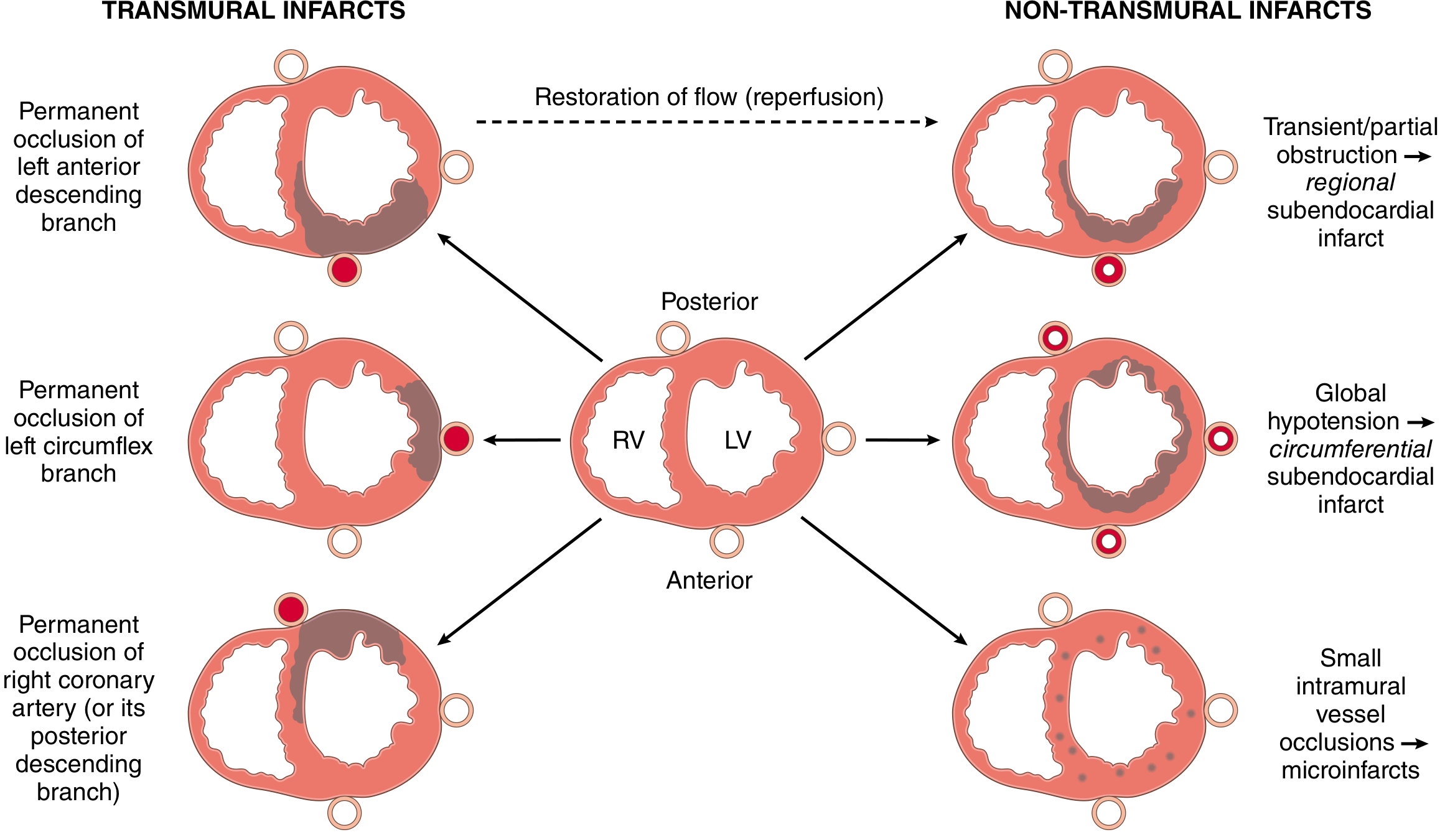
1. Transmural Infarction (STEMI)
- Full-thickness necrosis from subendocardium to epicardium
- Caused by complete occlusion of an epicardial coronary artery (atherosclerosis + thrombus)
- Associated with ST-elevation on ECG
2. Subendocardial Infarction (NSTEMI)
- Necrosis limited to the inner one-third to half of the myocardium
- Caused by: transient/partial coronary occlusion, global hypotension, severe fixed stenosis with prolonged hypoperfusion
- Circumferential subendocardial infarction occurs with global hypotension (not related to a single coronary territory)
- Not associated with complete coronary occlusion; often managed conservatively
3. Microinfarcts
- Caused by small intramural vessel occlusion (microembolization, vasculitis, vasospasm from catecholamines/cocaine)
Coronary Artery Territories (right-dominant circulation, ~80% of people)
| Artery | Frequency | Area Infarcted |
|---|---|---|
| LAD | 40-50% | Anterior LV wall, anterior 2/3 of interventricular septum, apex |
| RCA | 30-40% | Inferior/posterior LV wall, posterior 1/3 of septum, RV free wall (15-30% of RCA occlusions) |
| LCX | 15-20% | Lateral wall of LV (except apex) |
Morphological Changes (Gross and Microscopic Timeline)
This is the most exam-important section:
| Time | Gross Changes | Microscopic Changes |
|---|---|---|
| 0-30 min | None | None visible by light microscopy; ultrastructure shows mitochondrial swelling, glycogen depletion |
| 30 min - 4 hrs | None (EM: myofibrillar relaxation) | Wavy fibers at the periphery of the infarct |
| 4-12 hrs | Possible dark mottling | Early coagulative necrosis begins; edema, hemorrhage; neutrophil emigration begins at margins |
| 12-24 hrs | Dark mottling | Ongoing coagulative necrosis; pyknosis of nuclei; "hypereosinophilic" (deeply eosinophilic) myofibers; marginal contraction band necrosis (if reperfusion) |
| 1-3 days | Mottling with yellow-tan center | Coagulative necrosis fully established; intense neutrophilic infiltration |
| 3-7 days | Hyperemic border; central yellow-tan softening | Myocyte loss; macrophage infiltration begins; phagocytosis of dead cells |
| 7-10 days | Maximally yellow-tan and soft; depressed | Granulation tissue appears at margins; ingrowth of fibroblasts and vessels |
| 10-14 days | Red-gray depressed borders (early scar) | Established granulation tissue; collagen deposition starts |
| 2-8 weeks | Gray-white fibrosis progressively from margins | Increasing collagen deposition, decreasing cellularity |
| > 2 months | Dense white fibrous scar | Dense collagenous scar - complete healing |
Key point: An infarct achieves its full extent within 6-12 hours. Intervention in this critical window can limit the size.
Special gross stain: Triphenyl tetrazolium chloride (TTC) - viable myocardium stains brick-red; infarcted (necrotic) myocardium is unstained/pale (used for early infarct detection at autopsy).
Reperfusion Injury
When blood flow is restored, reperfusion can cause:
- Contraction band necrosis - hypercontracted sarcomeres appear as dense eosinophilic transverse bands across myofibers (caused by massive calcium influx into irreversibly damaged cells)
- Reperfusion arrhythmias - due to electrical instability
- Stunned myocardium - postischemic myocardium that is non-contractile for days despite being viable; causes transient reversible cardiac failure
- Hemorrhagic infarction - hemorrhage into the necrotic zone from damaged microvasculature
Despite these effects, early reperfusion is still beneficial overall - it salvages at-risk myocardium that hasn't yet undergone irreversible injury.
Cardiac Biomarkers
The earliest detectable feature of myocyte necrosis is disruption of the sarcolemmal membrane, allowing intracellular proteins to leak into the circulation.
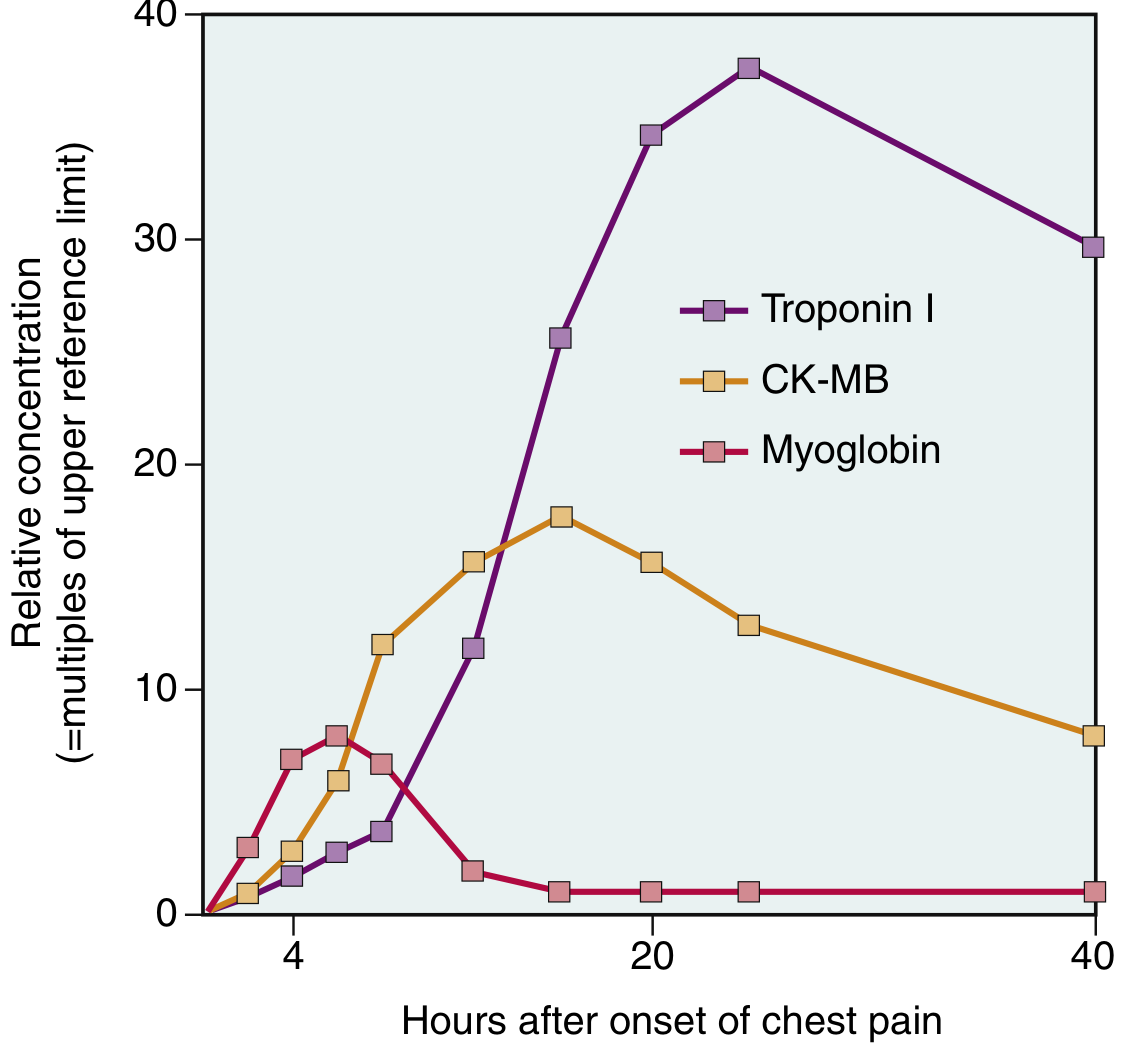
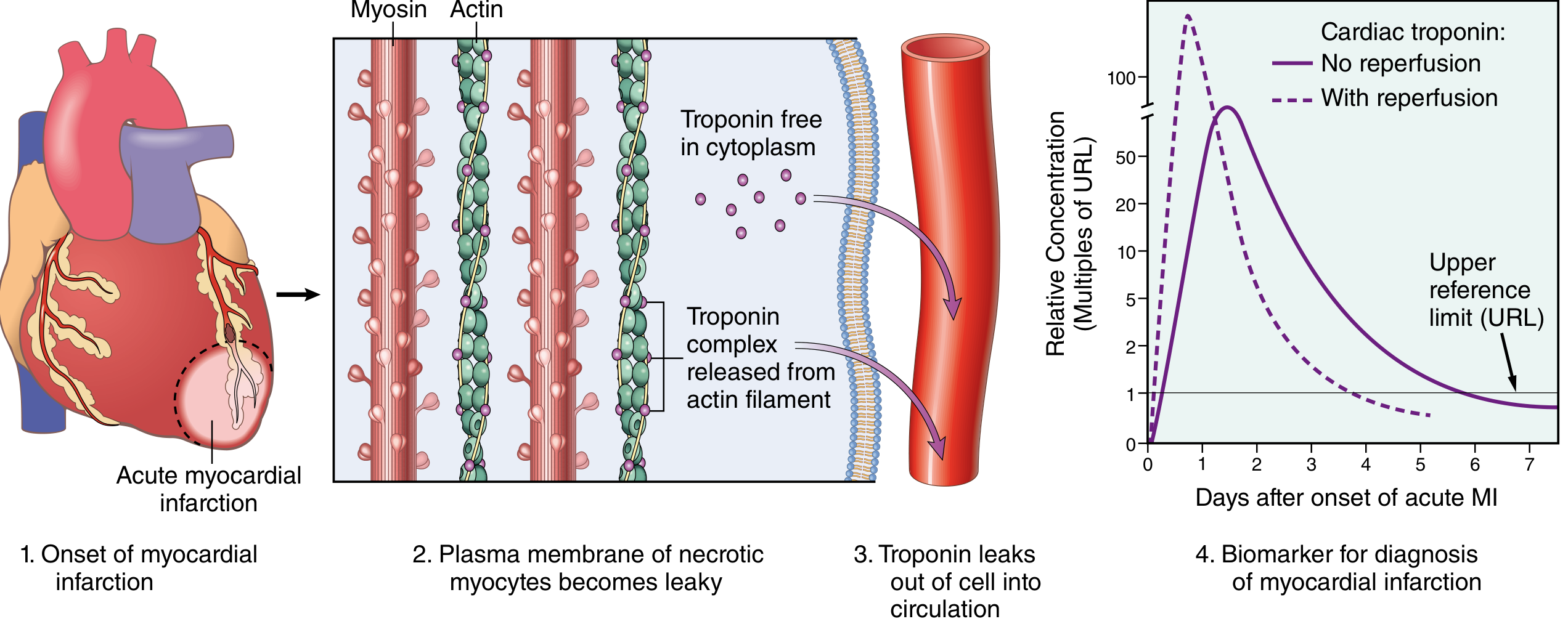
| Biomarker | Rise | Peak | Return to Normal |
|---|---|---|---|
| Myoglobin | 1-4 hrs (earliest) | 6-8 hrs | 24-48 hrs |
| CK-MB | 2-4 hrs | 24-48 hrs | ~72 hrs (3 days) |
| Troponin I/T | 2-4 hrs | 24-48 hrs | 7-10 days (most specific and sensitive) |
- Troponin I and T are the gold standard biomarkers - normally absent from circulation; highly specific for myocardial injury
- With reperfusion, both troponin and CK-MB peak earlier and higher due to washout from necrotic tissue
- Serial measurements help distinguish MI from other causes of troponin elevation (CHF, PE, sepsis, renal failure)
Complications of MI
Nearly three-fourths of patients experience one or more complications. Three are potentially lethal:
Cardiac Rupture (1-5% of MIs, frequently fatal)
- Ventricular septal rupture - most common site; produces a left-to-right shunt; VSD murmur
- Free wall rupture - catastrophic hemopericardium and cardiac tamponade; most common day 3-7 when the wall is softened by inflammation/necrosis but scar not yet formed
- Papillary muscle rupture - acute severe mitral regurgitation; more common with posterior papillary muscle (RCA territory, single blood supply)
Arrhythmias
- Most common cause of death in the early post-MI period
- In 80-90% of cases, cardiac death in MI setting is due to ventricular fibrillation
- Caused by electrical instability of ischemic myocardium
Left Ventricular Failure and Cardiogenic Shock
- Pump failure proportional to infarct size
- Cardiogenic shock when >40% of LV myocardium is infarcted
- Leads to pulmonary edema, hypotension
Pericarditis (Fibrinous/Epistenocardiac)
- Day 2-3 after transmural infarct; friction rub; chest pain
- Dressler syndrome (autoimmune pericarditis) - weeks to months later
Infarct Expansion and Ventricular Aneurysm
- Infarct expansion: acute dilatation and thinning of the necrotic wall (distinct from extension)
- True ventricular aneurysm: late complication; paradoxical systolic bulge; site for mural thrombus formation and systemic embolism
- Mural thrombus: forms on the endocardium overlying the infarcted area; risk of systemic embolism
Right Ventricular Infarction
- 15-30% of inferior MIs (RCA occlusion) extend into the RV
- Presents with right heart failure, elevated JVP, clear lungs
Extension of Infarction
- Enlargement of the original infarct zone due to ongoing ischemia
Clinical Features
- Chest pain: severe, crushing, retrosternal, radiating to left arm/jaw/neck; lasts >20 minutes
- Silent MI: especially in diabetics and elderly
- ECG changes: ST elevation (STEMI), Q waves (transmural), ST depression (NSTEMI/subendocardial)
- Biomarkers: troponin rise is diagnostic
Healing and Outcomes
- Overall in-hospital mortality: ~7-8%
- STEMI mortality: ~10%; NSTEMI mortality: ~6%
- Out-of-hospital STEMI mortality: ~33% (mostly from arrhythmia within 1 hour of onset)
- Scar formation is complete by 6-8 weeks in most transmural infarcts
- The fibrous scar is inert but does not contract - it impairs overall cardiac function
Content sourced from Robbins & Cotran Pathologic Basis of Disease (10th ed.) and Robbins Basic Pathology - the primary references underlying Harsh Mohan's Textbook of Pathology. The pathological descriptions and mechanisms are consistent across both texts.
This is a shared conversation. Sign in to Orris to start your own chat.