ETC cycle
Reading File
Finding Sources
Searching Images
electron transport chain ETC oxidative phosphorylation diagram
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a comprehensive overview of the Electron Transport Chain (ETC) based on Lippincott's Biochemistry and Basic Medical Biochemistry:
Electron Transport Chain (ETC)
Overview
The ETC is the final common pathway by which electrons derived from fuel molecules (carbohydrates, fats, amino acids) flow to oxygen. NADH and FADH₂ — generated by glycolysis, the TCA cycle, and β-oxidation — donate their electrons to the chain. As electrons pass through the complexes, they lose free energy, which is harnessed to pump H⁺ across the inner mitochondrial membrane, creating a proton gradient that drives ATP synthesis (oxidative phosphorylation / OXPHOS).
Location
Embedded in the inner mitochondrial membrane (except cytochrome c, which is in the intermembrane space). The inner membrane is impermeable to H⁺ and most small molecules — this impermeability is essential for maintaining the proton gradient.
The Four Complexes
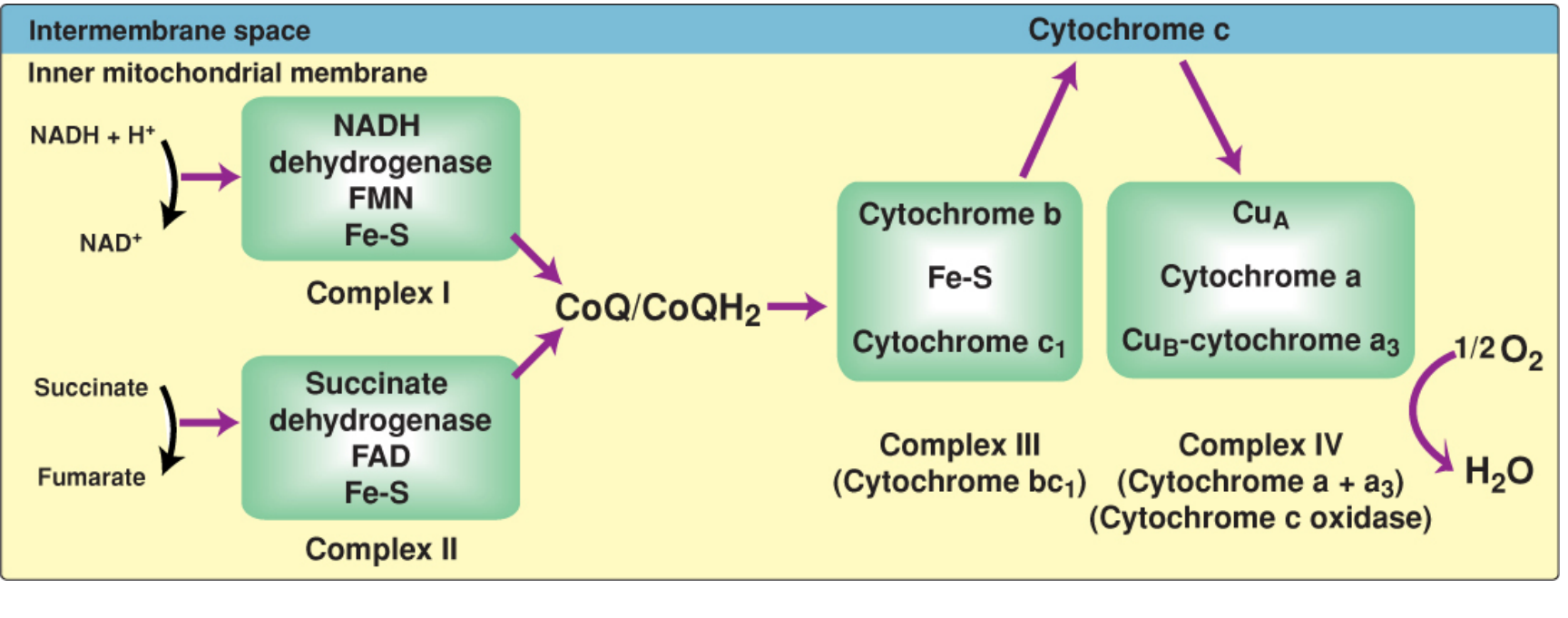
| Complex | Name | Cofactors | Electron donor → acceptor | H⁺ pumped |
|---|---|---|---|---|
| I | NADH dehydrogenase (NADH:CoQ oxidoreductase) | FMN, Fe-S centers | NADH → CoQ | 4 H⁺ |
| II | Succinate dehydrogenase | FAD, Fe-S centers | Succinate → CoQ (via FADH₂) | 0 |
| III | Cytochrome bc₁ (CoQ:cytochrome c oxidoreductase) | Cyt b, Fe-S, Cyt c₁ | CoQH₂ → Cytochrome c | 4 H⁺ |
| IV | Cytochrome c oxidase (Cyt a + a₃) | Cyt a, CuA, CuB, Cyt a₃ | Cytochrome c → O₂ → H₂O | 2 H⁺ |
Total H⁺ pumped per NADH: 10 (4+0+4+2, though Complex II bypasses the count)
Mobile Electron Carriers
- Coenzyme Q (CoQ / Ubiquinone): Lipid-soluble quinone. Collects electrons from Complexes I, II, and other flavoprotein dehydrogenases (glycerol 3-phosphate dehydrogenase, acyl-CoA dehydrogenase). Transfers to Complex III. Acts as a "hub."
- Cytochrome c: Small protein in intermembrane space, loosely associated with the outer face of the inner membrane. Shuttles electrons from Complex III → Complex IV.
Complex-by-Complex Detail
Complex I (NADH Dehydrogenase)
- Accepts 2e⁻ from NADH; FMN is reduced to FMNH₂
- Electrons pass through Fe-S centers → CoQ
- Pumps 4 H⁺ into intermembrane space
Complex II (Succinate Dehydrogenase)
- Catalyzes succinate → fumarate (TCA cycle step)
- FADH₂ → Fe-S → CoQ
- No H⁺ pumping (energy drop is insufficient)
Complex III (Cytochrome bc₁)
- CoQH₂ → Cyt b → Fe-S → Cyt c₁ → Cytochrome c
- Pumps 4 H⁺ via the Q cycle
- Electrons pass as single electrons (CoQ carries 2e⁻, cytochromes carry 1e⁻)
Complex IV (Cytochrome c Oxidase)
- CuA → Cyt a → CuB-Cyt a₃ → O₂
- Only complex that directly reacts with O₂
- Reduces O₂ to H₂O (requires 4 electrons + 4H⁺)
- Pumps 2 H⁺ per pair of electrons
ATP Yield
| Electron donor | Entry point | ATP yield (approx.) |
|---|---|---|
| NADH | Complex I | ~2.5 ATP |
| FADH₂ | Complex II (via CoQ) | ~1.5 ATP |
Complete oxidation of 1 glucose → ~30–32 ATP total.
ETC Inhibitors
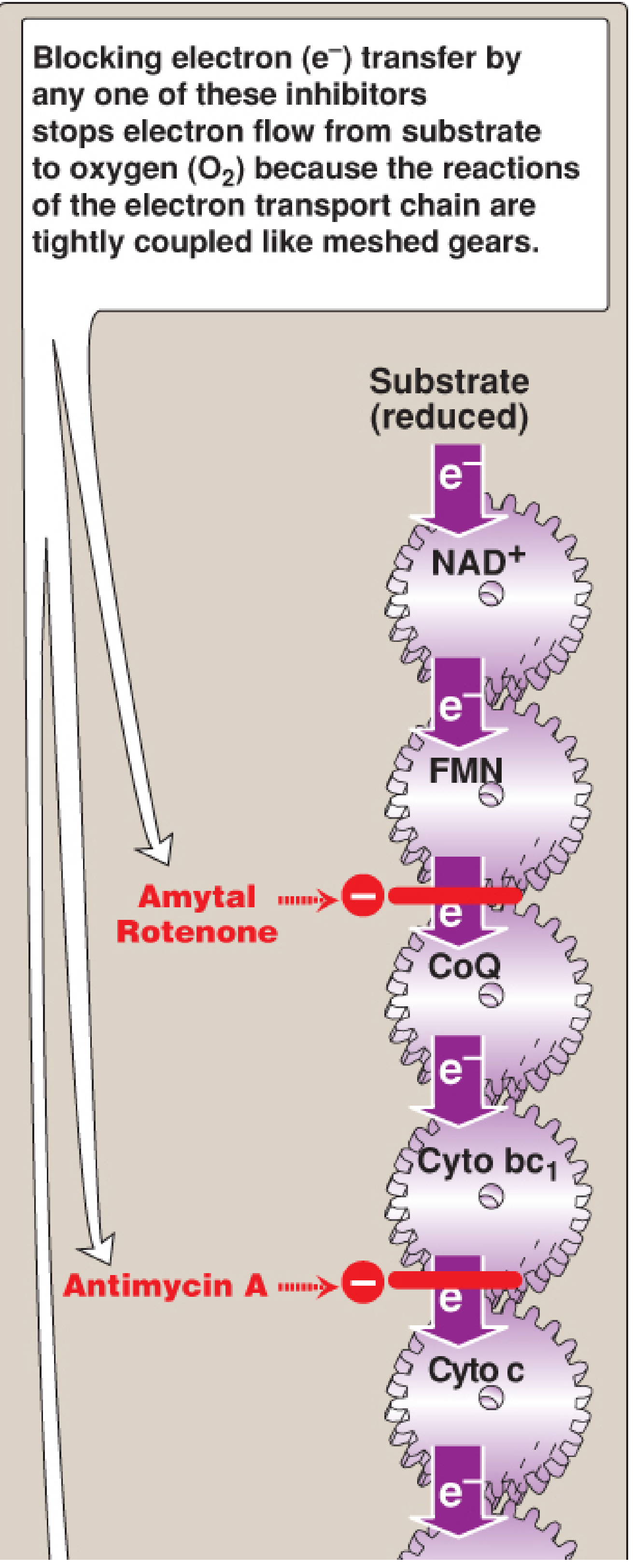
| Inhibitor | Site blocked | Mechanism |
|---|---|---|
| Rotenone / Amytal | Complex I | Block FMN → CoQ transfer |
| Antimycin A | Complex III | Block Cyt bc₁ → Cyt c |
| Cyanide (CN⁻), CO, H₂S, NaN₃ | Complex IV | Bind heme Fe of Cyt a₃; prevent O₂ reduction |
Blocking any single complex halts the entire chain because the reactions are tightly coupled ("meshed gears" — if one stops, all stop).
Reactive Oxygen Species (ROS)
Electron leakage from Complexes I and III can reduce O₂ to superoxide (O₂⁻). This leads to:
- H₂O₂, hydroxyl radicals (•OH)
- DNA damage, lipid peroxidation, protein oxidation
Cellular defenses: Superoxide dismutase (SOD), catalase, glutathione peroxidase.
Cytoplasmic NADH Shuttles
Since NADH cannot cross the inner mitochondrial membrane, two shuttle systems transfer its reducing equivalents:
| Shuttle | Tissue | Enters chain as | ATP yield |
|---|---|---|---|
| Glycerol 3-phosphate shuttle | Most tissues (muscle, brain) | FADH₂ (via CoQ) | ~1.5 ATP |
| Malate-aspartate shuttle | Heart, liver | NADH (Complex I) | ~2.5 ATP |
Key clinical point: Ischemia (O₂ deprivation) stops the ETC — without O₂ as the terminal electron acceptor, H⁺ pumping ceases, the proton gradient collapses, and ATP synthesis halts, leading to cell injury.
Sources: Biochemistry, 8th ed. Lippincott Illustrated Reviews, pp. 224–236 | Basic Medical Biochemistry, 6th ed., pp. 868–874
Ureae cycle
Finding Sources
Searching Images
urea cycle diagram steps enzymes
Reading File
Reading File
Loading Image
Reading File
Reading File
Loading Image
Reading File
Here is a comprehensive overview of the Urea Cycle drawn from Lippincott's Biochemistry and Basic Medical Biochemistry:
Urea Cycle (Krebs-Henseleit Cycle)
Overview
Urea (H₂N-CO-NH₂) is the major disposal form of amino nitrogen in the body, accounting for ~90% of nitrogen excreted in urine. The cycle was first proposed in 1932 by Hans Krebs and Kurt Henseleit.
- N₁ of urea → from free NH₃ (via GDH-mediated deamination of glutamate)
- N₂ of urea → from aspartate
- Carbon & oxygen → from CO₂ (as HCO₃⁻)
- Produced exclusively in the liver (arginase-I is liver-specific)
Compartmentalization
| Steps | Location |
|---|---|
| Steps 1–2 (carbamoyl phosphate → citrulline) | Mitochondrial matrix |
| Steps 3–5 (citrulline → urea) | Cytosol |
Citrulline exits and ornithine enters the mitochondria via an antiporter (ORNT1).
The 5 Steps
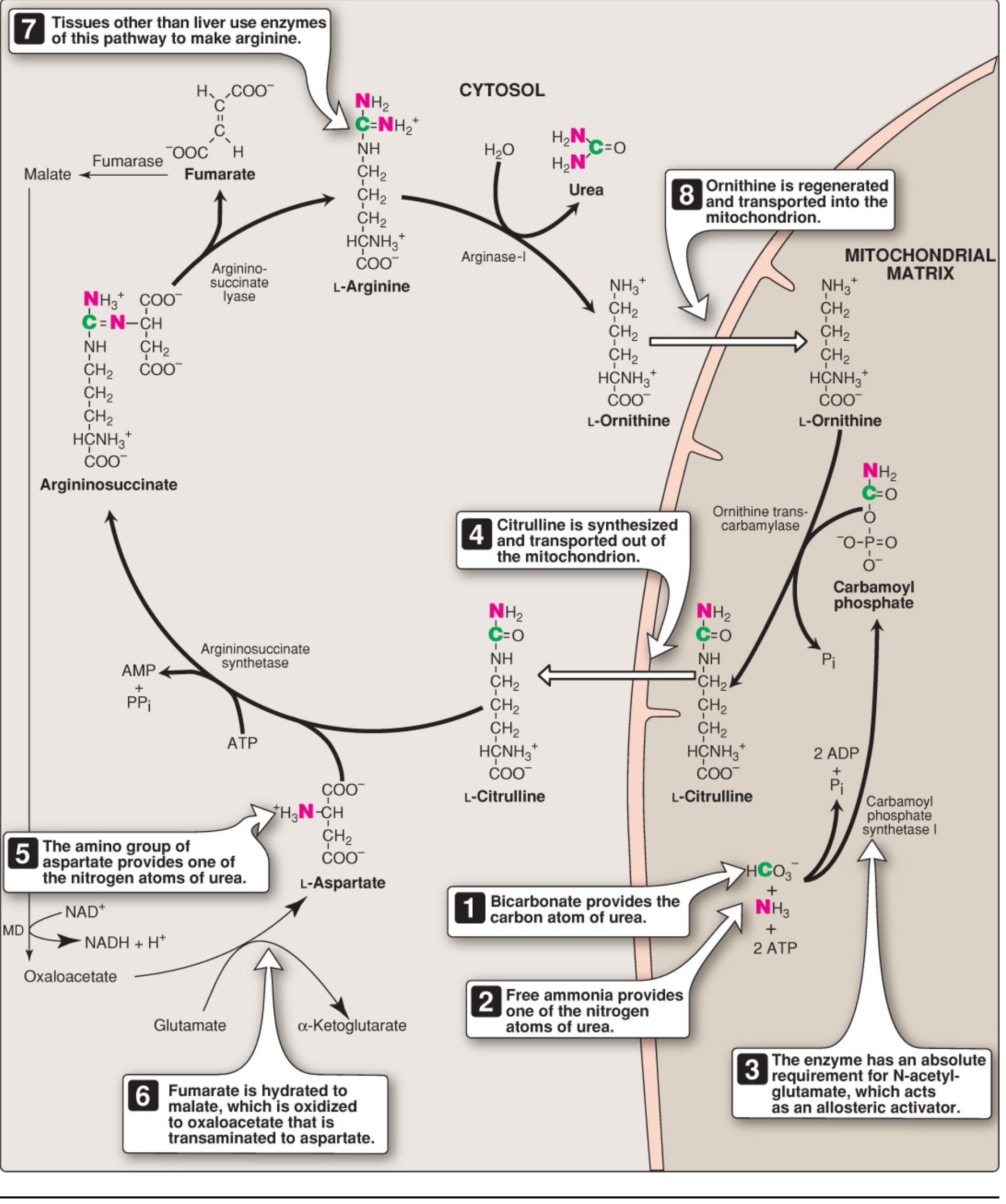
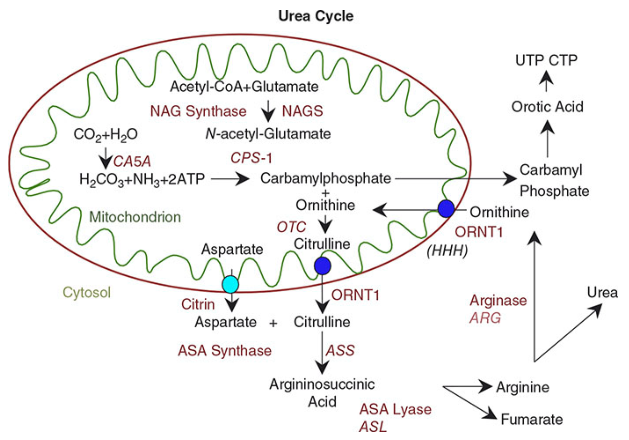
| Step | Reaction | Enzyme | Location | ATP cost | Notes |
|---|---|---|---|---|---|
| 1 | NH₃ + HCO₃⁻ + 2ATP → Carbamoyl phosphate | CPS-I (Carbamoyl Phosphate Synthetase I) | Mitochondria | 2 ATP | Requires N-acetylglutamate (NAG) as allosteric activator |
| 2 | Carbamoyl phosphate + Ornithine → Citrulline | OTC (Ornithine TransCarbamylase) | Mitochondria | — | Citrulline exported to cytosol via ORNT1 |
| 3 | Citrulline + Aspartate → Argininosuccinate | Argininosuccinate Synthetase (ASS) | Cytosol | 1 ATP → AMP + PPi | Aspartate donates the 2nd nitrogen |
| 4 | Argininosuccinate → Arginine + Fumarate | Argininosuccinate Lyase (ASL) | Cytosol | — | Fumarate enters TCA or gluconeogenesis |
| 5 | Arginine + H₂O → Ornithine + Urea | Arginase-I | Cytosol | — | Liver-specific; ornithine recycled back |
Overall Stoichiometry
Aspartate + NH₃ + HCO₃⁻ + 3 ATP + H₂O → Urea + Fumarate + 2 ADP + AMP + 2 Pᵢ + PPᵢ
- 4 high-energy phosphate bonds consumed per molecule of urea (2 ATP at CPS-I → 2 ADP; 1 ATP at ASS → AMP + PPi = 2 bonds)
- Reaction is irreversible (large negative ΔG)
Regulation
| Regulator | Effect | Notes |
|---|---|---|
| N-Acetylglutamate (NAG) | ↑ CPS-I activity (allosteric activator) | Rate-limiting step; NAG is made from acetyl-CoA + glutamate by NAG Synthase (NAGS) |
| Arginine | Activates NAGS → ↑ NAG → ↑ CPS-I | Positive feedback |
| Protein intake | Long-term enzyme induction | High-protein diet upregulates all urea cycle enzymes |
| Substrate availability | Short-term regulation | Flux driven by NH₃ and aspartate supply |
Link to Other Pathways
The urea cycle is tightly linked to the TCA cycle via the fumarate–malate–oxaloacetate–aspartate loop:
Fumarate (from Step 4)
↓ Fumarase
Malate
↓ Malate dehydrogenase
Oxaloacetate
↓ Transaminase (AST)
Aspartate → re-enters urea cycle at Step 3
This is sometimes called the "Krebs bicycle." Fumarate carbons can also feed gluconeogenesis.
Ammonia Transport to the Liver
Ammonia is toxic (especially to the CNS). Peripheral tissues transport nitrogen safely to the liver as:
| Carrier | Tissue of origin | Mechanism |
|---|---|---|
| Glutamine | Muscle, brain | Glutamine synthetase adds NH₃ to glutamate; liver glutaminase releases it |
| Alanine | Muscle | Glucose-alanine cycle; ALT transfers NH₃ to pyruvate → alanine |
Urea Cycle Enzyme Defects (Key Disorders)
| Deficiency | Enzyme | Finding | Key clue |
|---|---|---|---|
| CPS-I deficiency | CPS-I | Hyperammonemia | No orotic aciduria |
| OTC deficiency | OTC | Hyperammonemia | ↑ Urinary orotic acid (carbamoyl phosphate spills into pyrimidine synthesis) — most common urea cycle disorder; X-linked |
| Citrullinemia | ASS | ↑ Citrulline | Hyperammonemia |
| Argininosuccinic aciduria | ASL | ↑ Argininosuccinate in urine | Hyperammonemia |
| Argininemia | Arginase-I | ↑ Arginine | Spastic diplegia, less severe hyperammonemia |
| NAGS deficiency | NAGS | Hyperammonemia | No orotic aciduria (mimics CPS-I) |
OTC deficiency is the most common and is X-linked; heterozygous females may have episodic hyperammonemia.
Clinical: Hyperammonemia
- Symptoms: vomiting, lethargy, cerebral edema, encephalopathy, coma
- Treatment: low-protein diet, arginine supplementation, sodium benzoate/phenylbutyrate (alternative nitrogen disposal), liver transplant (definitive)
Sources: Biochemistry, 8th ed. Lippincott Illustrated Reviews, pp. 712–718 | Basic Medical Biochemistry, 6th ed., pp. 1321–1323
This is a shared conversation. Sign in to Orris to start your own chat.