Respiratory system pathology Chronic diffuse interstitial disease Idiopathic pulmonary fibrosis etc Add pictures of gross, microscopy, add mnemonic if possible
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Loading Image
Loading Image
Finding Sources
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Reading File
Loading Image
I now have comprehensive content from Robbins Pathology, Harrison's, Murray & Nadel, and Fishman's. Let me now compile the full, detailed answer:
Chronic Diffuse Interstitial (Restrictive) Lung Diseases
Overview & Classification
Chronic diffuse interstitial lung diseases (ILDs) are a heterogeneous group of bilateral, often patchy pulmonary disorders characterized by fibrosis of alveolar walls. They share common features: reduced lung compliance (stiff lungs), dyspnea, restrictive PFTs, and ground-glass or reticular opacities on CXR/CT. In advanced stages, all lead to the same endpoint - "honeycomb lung."
Categories (Robbins Basic Pathology, Table 11.2):
| Category | Examples |
|---|---|
| Fibrosing | IPF/UIP, NSIP, COP, Collagen vascular disease, Pneumoconiosis |
| Granulomatous | Sarcoidosis, Hypersensitivity pneumonia |
| Eosinophilic | Loeffler syndrome, Idiopathic eosinophilic pneumonia |
| Smoking-related | DIP, Respiratory bronchiolitis-ILD |
🔑 MNEMONIC: "FGSE" for ILD Categories
Fibrosing, Granulomatous, Smoking-related, Eosinophilic
IDIOPATHIC PULMONARY FIBROSIS (IPF) / Usual Interstitial Pneumonia (UIP)
Definition
IPF is a pulmonary disorder of unknown etiology characterized by patchy, progressive bilateral interstitial fibrosis. The histologic/radiologic pattern is called Usual Interstitial Pneumonia (UIP), which is required for the diagnosis. It is also known as cryptogenic fibrosing alveolitis. IPF is a diagnosis of exclusion - similar UIP patterns can occur in asbestosis, collagen vascular disease, etc.
Epidemiology
- Prevalence: 50-200 per 100,000
- Predominantly affects males > females
- Typically diagnosed in the 5th-6th decade (virtually never before age 50)
- Associated with smoking, air pollution, metal fumes, wood dust, farming, hairdressing, stone polishing
- Poor prognosis: 50% survival at 3-5 years
Pathogenesis
IPF arises from repeated alveolar epithelial injury in genetically predisposed individuals.
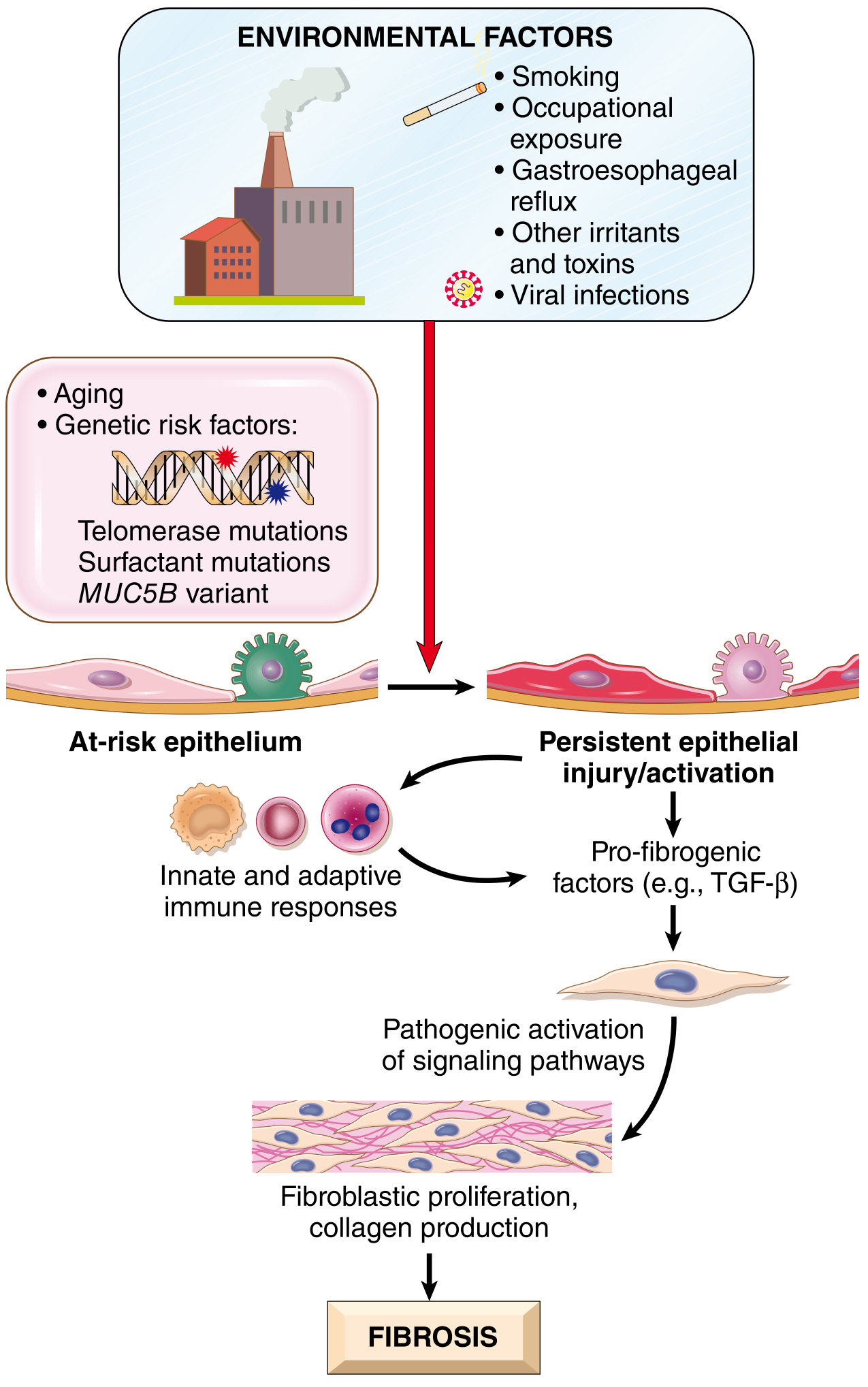
Environmental Triggers
- Cigarette smoking (greatest risk factor - increases risk several-fold)
- Air pollution, microaspiration, metal fumes, wood dust
- Farming, hairdressing, stone polishing occupations
Genetic Factors
- TERT, TERC, PARN, RTEL1 mutations (telomere maintenance genes) - up to 15% of familial IPF
- Surfactant gene mutations - cause protein misfolding → unfolded protein response → type II pneumocyte sensitization
- MUC5B promoter SNP - greatly increases MUC5B mucin secretion (alters mucociliary clearance)
- Up to 25% of sporadic IPF: abnormal telomere shortening in peripheral blood lymphocytes
The Fibrotic Cascade
- Persistent alveolar epithelial injury/activation
- Injured type II pneumocytes release TGF-β and other profibrotic factors
- Innate/adaptive immune cells amplify the signal
- Fibroblast/myofibroblast activation and proliferation
- Excessive collagen deposition → interstitial fibrosis
🔑 MNEMONIC for IPF Risk Factors: "SAGE-TOM"
Smoking, Aging, Genetics (telomerase/MUC5B), Environmental exposure (dust/fumes) - Telomere shortening, Occupational (farming/stone polishing), Microaspiration (GERD)
Gross Pathology
- Pleural surfaces are cobblestoned due to retraction of scars along interlobular septa
- Cut surface: firm, rubbery, white areas of fibrosis, predominantly at the periphery/bases
- Subpleural and basilar predominance is characteristic
- End-stage: Honeycomb lung - dilated, cystic spaces resembling a honeycomb
Microscopy (Histopathology) - UIP Pattern
The hallmark features are:
| Feature | Description |
|---|---|
| Temporal heterogeneity | Old dense fibrosis AND fresh fibroblastic foci coexist in the same biopsy |
| Spatial heterogeneity | Fibrosis alternates with areas of preserved normal lung |
| Fibroblastic foci | Subepithelial collections of myofibroblasts and loose collagen - earliest lesion |
| Honeycomb fibrosis | Cystic spaces from collapsed alveolar walls, lined by hyperplastic type II pneumocytes or bronchiolar epithelium |
| Subpleural/paraseptal | Predominantly subpleural and adjacent to septa |
| Inflammation | Mild lymphocytic/plasma cell infiltrate in alveolar septa |
| Pulmonary HTN changes | Intimal fibrosis and medial thickening of pulmonary arteries (secondary) |
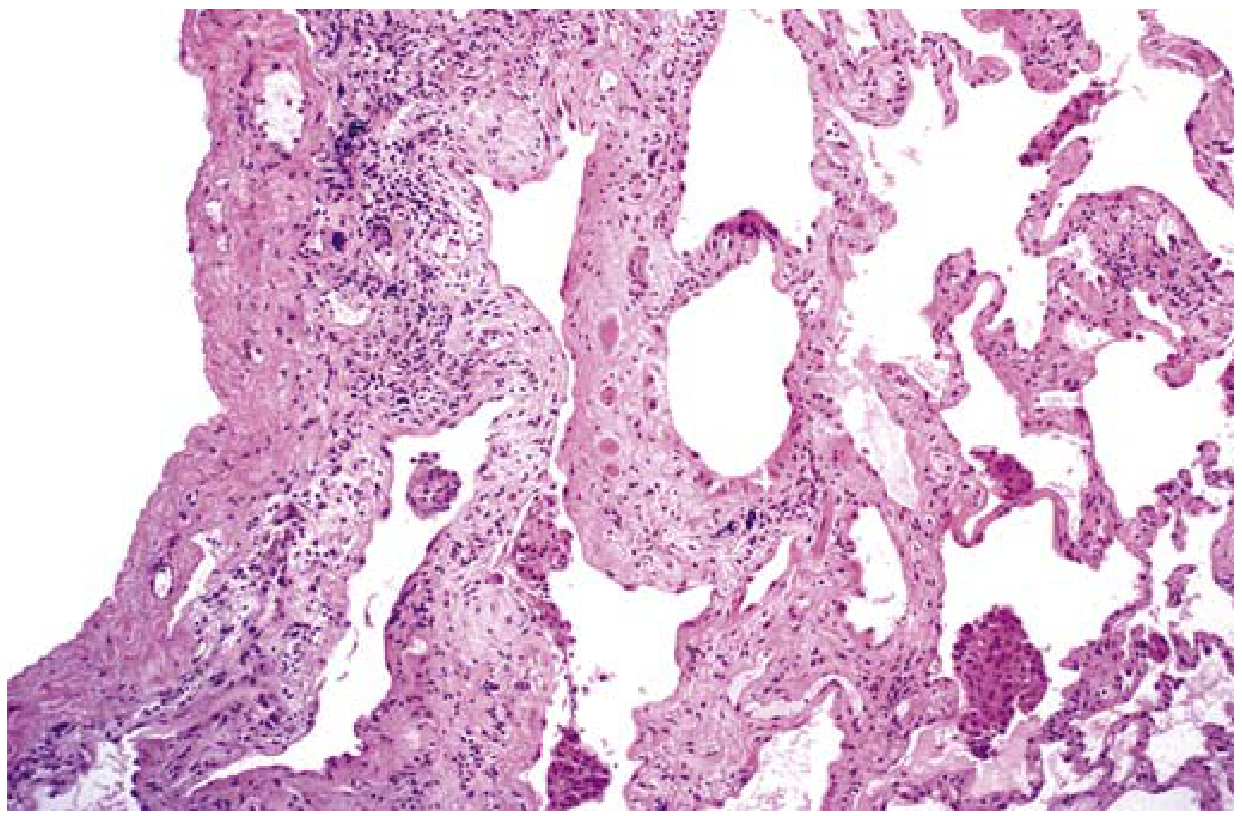
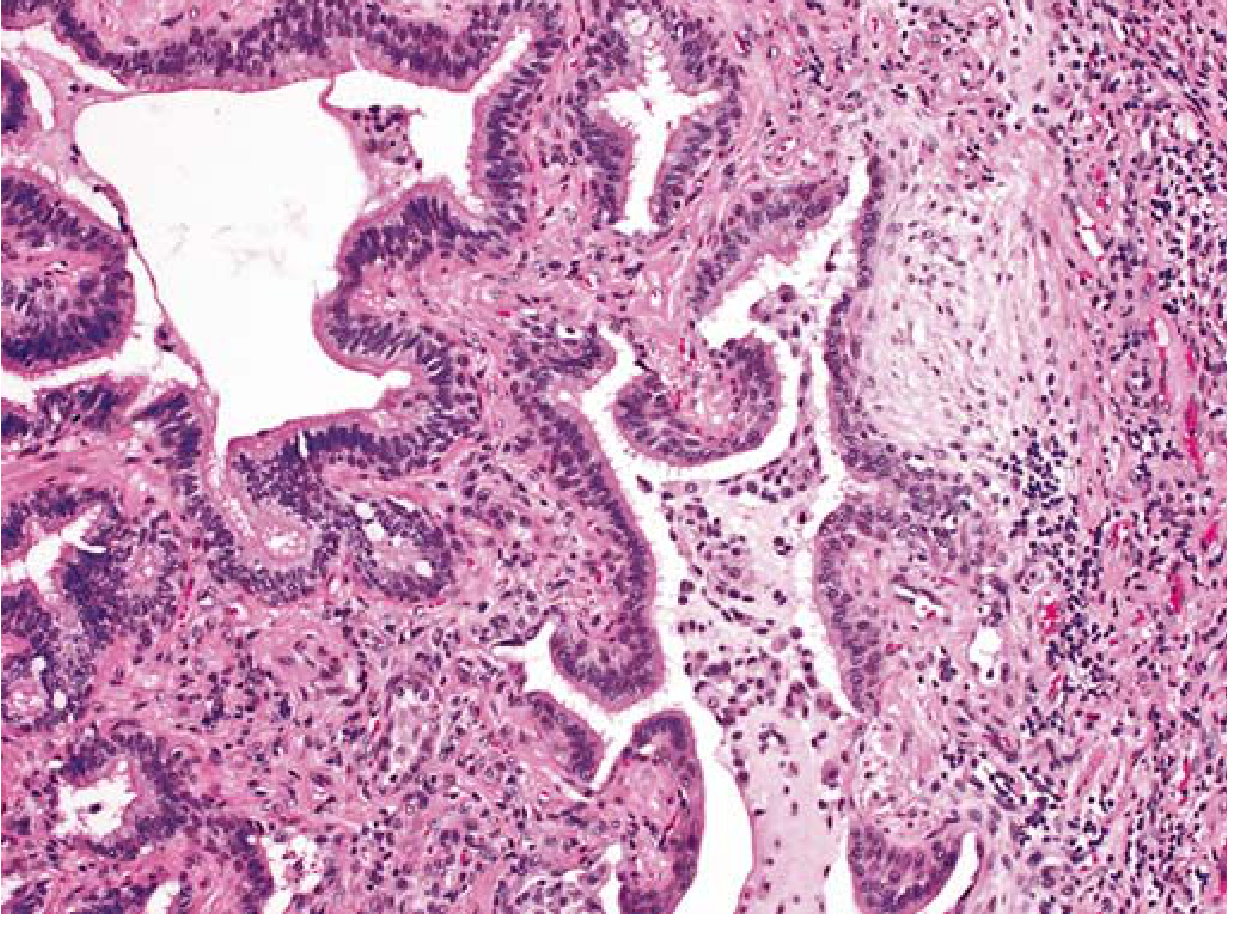
🔑 MNEMONIC for UIP Microscopy: "FISH-TH"
Fibroblastic foci, Inflammation (lymphocytes/plasma cells), Spatial heterogeneity, Honeycombing - Temporal heterogeneity, Hyperplastic type II pneumocytes lining cysts
Radiology (HRCT) - UIP Pattern
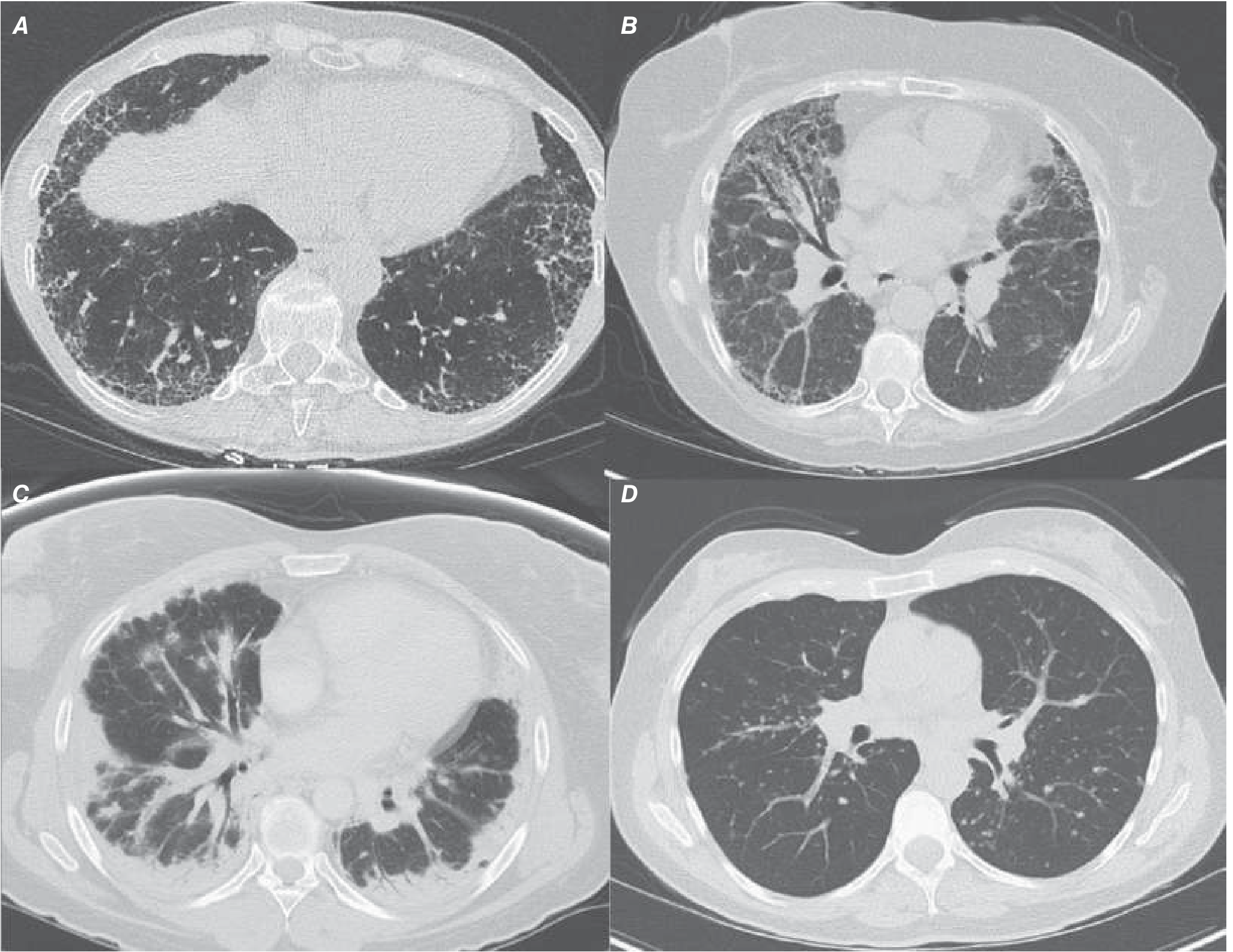
Classic UIP on HRCT:
- Posterior, basilar, subpleural predominance
- Reticular markings (irregular fibrotic lines)
- Traction bronchiectasis (airway distortion from surrounding fibrosis)
- Honeycombing (cystic spaces in rows, subpleural)
Features that argue against IPF: upper lung predominance, extensive ground-glass, micronodules, mosaic attenuation, bronchovascular changes
Histopathology Comparison - ILDs
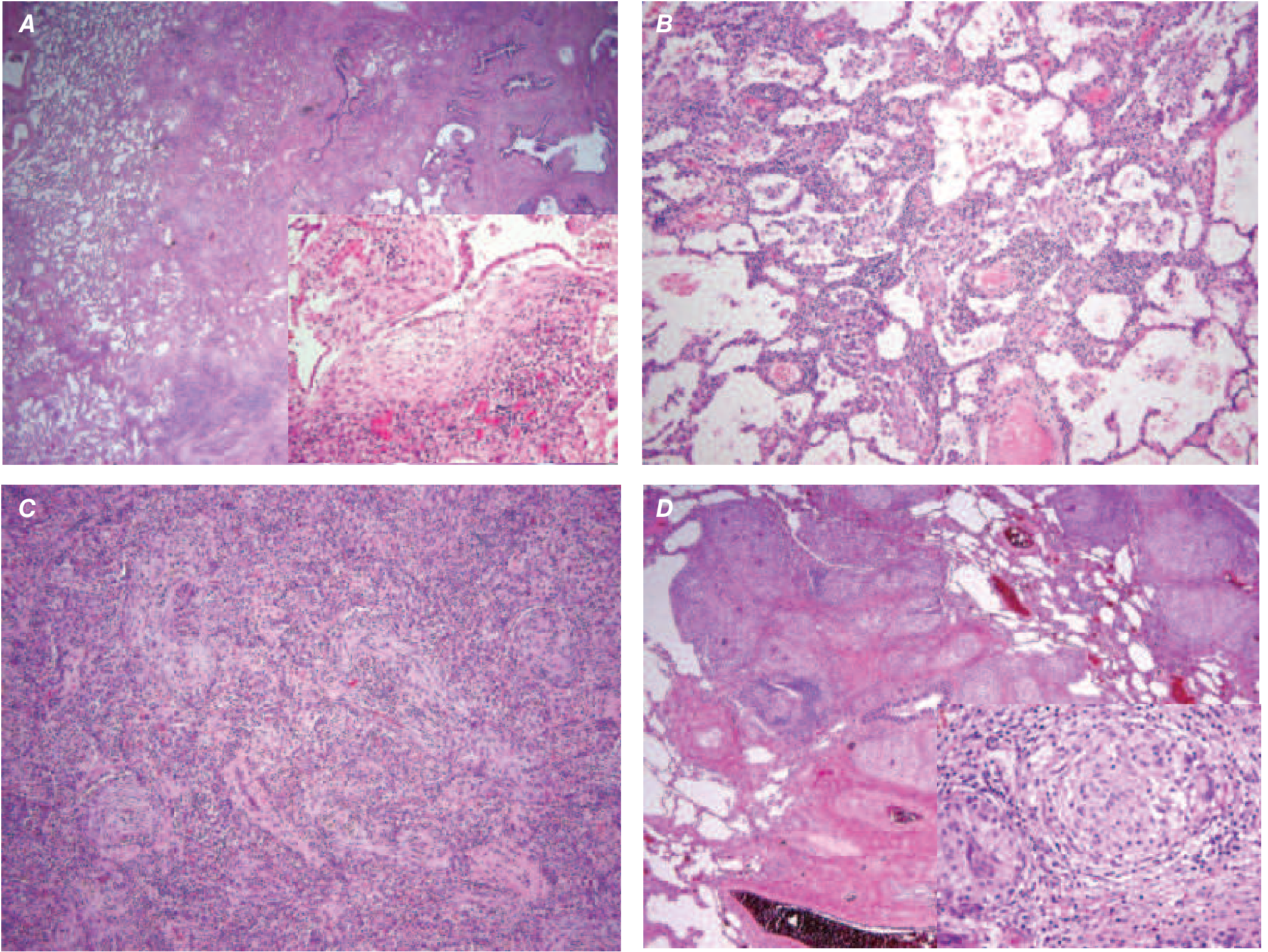
Clinical Features
- Gradual onset of non-productive cough + progressive exertional dyspnea
- "Velcro" or "dry" bibasilar crackles on auscultation (characteristic)
- Digital clubbing (in ~50%)
- Late: cyanosis, cor pulmonale, peripheral edema
- Pulmonary function tests: restrictive pattern with reduced DLCO
Treatment
- Antifibrotic therapy: Pirfenidone (TGF-β inhibitor) and Nintedanib (tyrosine kinase inhibitor) - both slow the rate of FVC decline, approved since 2014
- Immunosuppression (steroids/azathioprine/NAC) - shown to increase morbidity and mortality in IPF; NOT recommended
- Supplemental O₂ + pulmonary rehabilitation for exercise tolerance
- Lung transplantation - only definitive treatment; extends survival
OTHER KEY CHRONIC DIFFUSE INTERSTITIAL DISEASES
Nonspecific Interstitial Pneumonia (NSIP)
| Feature | Description |
|---|---|
| Who | Non-smoking females, 5th decade; often CTD-associated (RA, SLE, scleroderma) |
| Histology | Uniform interstitial inflammation +/- fibrosis (no temporal heterogeneity); absent honeycombing; rare fibroblastic foci |
| HRCT | Bilateral symmetric ground-glass + reticular; lower lobe; subpleural sparing (distinctive); bronchovascular thickening |
| Prognosis | Much better than IPF: >80% 5-year survival |
| Treatment | Steroids, mycophenolate, azathioprine, cyclophosphamide, rituximab |
Key distinction from IPF: NSIP shows temporal homogeneity (all lesions look the same age) vs. IPF's temporal heterogeneity (old fibrosis + fresh foci together).
Cryptogenic Organizing Pneumonia (COP)
| Feature | Description |
|---|---|
| Histology | Intra-alveolar plugs of loose organizing connective tissue (Masson bodies) in alveolar ducts + small airways; surrounding alveolar wall inflammation |
| HRCT | Patchy subpleural consolidation (migratory!); "reversed halo / atoll sign" (rim of consolidation around ground-glass) |
| Treatment | Oral steroids - often very responsive; some resolve spontaneously |
| Prognosis | Generally favorable |
Smoking-Related ILD
Respiratory Bronchiolitis-ILD (RB-ILD)
- Active heavy smokers, age 40-50
- Histology: pigmented macrophages in respiratory bronchioles and alveolar ducts (peribronchiolar)
- HRCT: centrilobular nodules, ground-glass, bronchial wall thickening
- Treatment: Smoking cessation (first and most important step)
Desquamative Interstitial Pneumonia (DIP)
- More diffuse than RB-ILD
- Histology: diffuse alveolar filling with pigmented macrophages + pneumocyte hyperplasia + prominent interstitial thickening
- HRCT: diffuse/patchy bilateral symmetric ground-glass opacities
- Treatment: smoking cessation ± steroids
Sarcoidosis (Granulomatous ILD)
| Feature | Description |
|---|---|
| Histology hallmark | Non-caseating granulomas (epithelioid cells + multinucleated giant cells, no central necrosis); peribronchiolar distribution along lymphatics |
| HRCT | Bilateral hilar lymphadenopathy + peribronhovascular nodules; upper/mid lung predominance |
| Pathogenesis | TGF-β, fibronectin, IGF-1 from alveolar macrophages → fibrosis in chronic cases |
| Advanced disease | Can progress to honeycombing; complications include bronchiectasis, mycetoma, hemoptysis |
Pulmonary Langerhans Cell Histiocytosis (PLCH)
- Young male smokers (2nd-3rd decade)
- Histology: interstitial Langerhans cells with Birbeck granules (X bodies on EM); CD1a+, S-100+ on IHC; peribronchiolar stellate fibrosis + cysts
- HRCT: upper/mid lung cysts + nodules; spares lung bases
- Somatic BRAF V600E mutations in many cases
- Treatment: smoking cessation; corticosteroids; vemurafenib (BRAF V600E+)
- Spontaneous pneumothorax in ~25%
Summary Comparison Table
| Disease | Histologic Pattern | Key Microscopic Feature | HRCT | Prognosis |
|---|---|---|---|---|
| IPF | UIP | Fibroblastic foci + temporal heterogeneity + honeycombing | Basal subpleural reticulation + honeycombing | Poor (3-5 yr survival) |
| NSIP | NSIP | Uniform fibrosis/inflammation, no honeycombing | GGO + reticulation; subpleural sparing | Good (>80% 5-yr) |
| COP | Organizing pneumonia | Masson bodies (intra-alveolar plugs) | Migratory consolidation; atoll sign | Good (steroid responsive) |
| DIP | DIP | Diffuse alveolar macrophages | Diffuse GGO | Moderate |
| Sarcoidosis | Granulomatous | Non-caseating granulomas | BHL + peribronhovascular nodules | Variable |
| PLCH | Langerhans cell | Birbeck granules; stellate fibrosis | Upper cysts + nodules | Variable |
🔑 Master Mnemonic - ILD Quick Review: "IPF needs PIRFENIDONE - Fibrosis Is Scary History"
Fibroblastic foci = earliest lesion
Interstitial inflammation (lymphocytes)
Spatial heterogeneity (alternating normal/fibrotic)
Honeycombing (late, end-stage)
Temporal heterogeneity (old + new lesions together)
Hyperplastic type II pneumocytes line honeycomb cysts
Sources: - Robbins Basic Pathology, Chapter 11 | Robbins & Kumar Pathologic Basis of Disease, Chapter 15 | Harrison's Principles of Internal Medicine 22E, Chapter 304 | Murray & Nadel's Textbook of Respiratory Medicine | Fishman's Pulmonary Diseases and Disorders
This is a shared conversation. Sign in to Orris to start your own chat.