Acetaminophen toxicity pathophysiology how it occur why it occurs in detail
Reading File
Finding Sources
Finding Sources
Searching PubMed
acetaminophen toxicity pathophysiology NAPQI
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive data from multiple major textbooks plus a clear metabolic pathway diagram. I have everything needed for a detailed answer.
Acetaminophen (Paracetamol) Toxicity — Detailed Pathophysiology
1. Normal Metabolism at Therapeutic Doses
At standard doses, acetaminophen is absorbed rapidly (peak concentration within 1–2 hours). The liver processes it via three parallel pathways:
| Pathway | Enzyme | Product | % of dose |
|---|---|---|---|
| Glucuronidation | UDP-glucuronosyltransferase | Acetaminophen-glucuronide (non-toxic) | 40–67% |
| Sulfation | Phenolsulfotransferase | Acetaminophen-sulfate (non-toxic) | 20–46% |
| CYP450 oxidation | CYP2E1 (primary), CYP1A2, CYP3A4 | NAPQI (toxic) | 5–15% |
The glucuronide and sulfate conjugates are water-soluble and safely excreted in urine. The small fraction oxidized by CYP2E1 produces N-acetyl-p-benzoquinone imine (NAPQI) — a highly reactive, electrophilic metabolite. At therapeutic doses, NAPQI is rapidly neutralized by hepatic glutathione (GSH) via glutathione-S-transferase, forming a non-toxic mercapturate conjugate (APAP-mercaptate), which is then excreted renally.
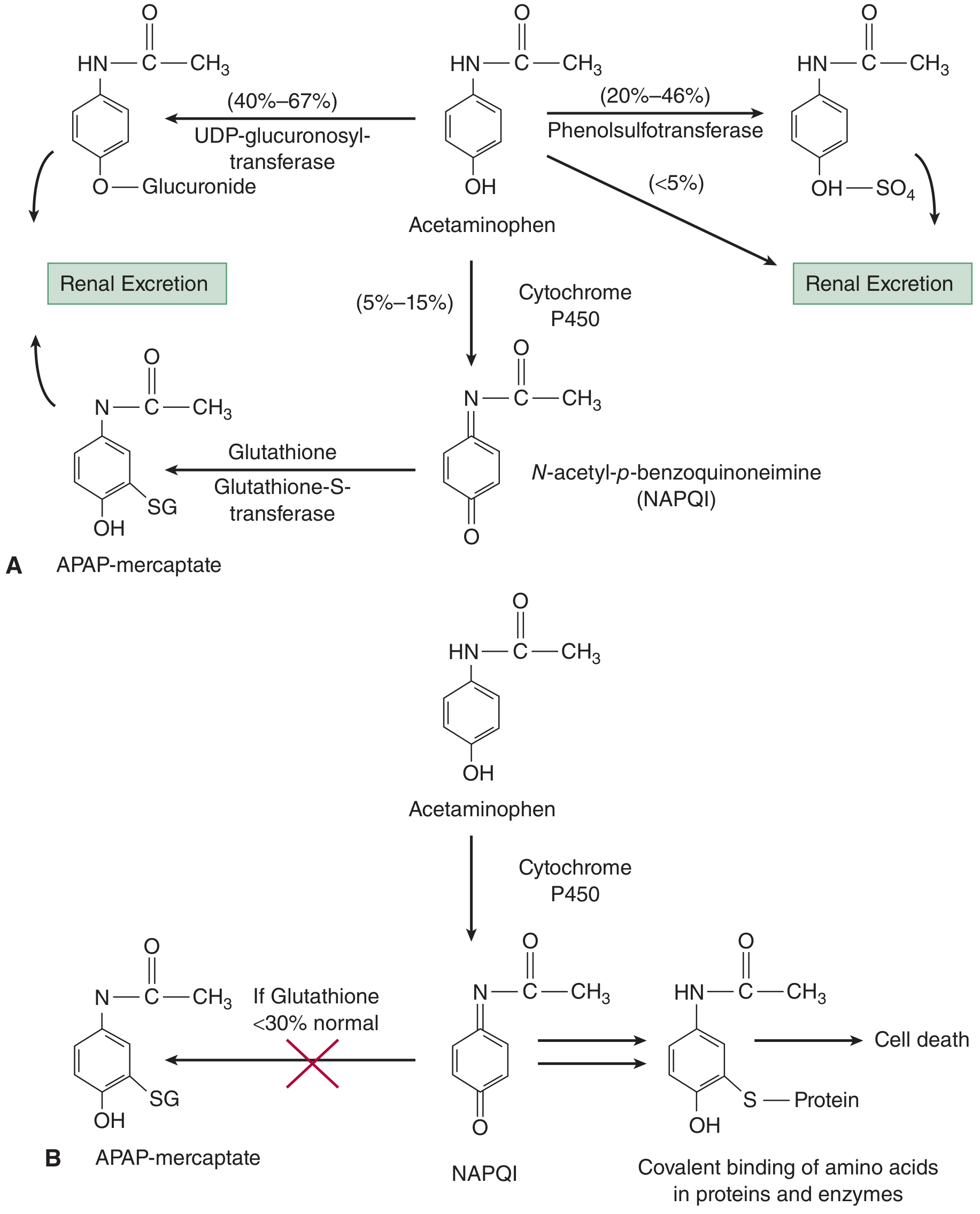
2. Why Toxicity Occurs — The Core Mechanism
Step 1: Saturation of Safe Pathways
After a large ingestion (typically >7.5–10 g in adults, or >150 mg/kg), the glucuronidation and sulfation pathways become saturated. As a result, a disproportionately larger fraction of acetaminophen is shunted through the CYP2E1 pathway, producing far more NAPQI than normal.
Step 2: Glutathione Depletion
The liver has finite glutathione stores. Under normal conditions, GSH is regenerated fast enough to handle small amounts of NAPQI. But with massive NAPQI production, hepatic GSH is consumed faster than it can be regenerated. When GSH falls below ~30% of normal, the detoxification capacity is overwhelmed.
Step 3: Covalent Binding → Hepatocyte Death
Unbound NAPQI is an extremely reactive electrophile. It covalently binds to sulfhydryl (–SH) groups on hepatocyte macromolecules — proteins, enzymes, and nucleic acids — forming acetaminophen-protein adducts. This triggers:
- Mitochondrial dysfunction — NAPQI disrupts the electron transport chain, reduces ATP synthesis, and increases reactive oxygen species (ROS)
- Oxidative stress — excess ROS causes lipid peroxidation and further protein damage
- Calcium dysregulation — loss of mitochondrial integrity disrupts intracellular Ca²⁺ homeostasis
- Hepatocyte necrosis — the combined insults lead to cell death by necrosis (not apoptosis)
- Inflammatory amplification — dying hepatocytes release damage-associated molecular patterns (DAMPs) that recruit neutrophils and Kupffer cells, amplifying liver destruction
Step 4: Zone III (Centrilobular) Predominance
Hepatic damage is concentrated in hepatic Zone III (centrilobular) because:
- CYP2E1 enzyme concentration is highest around the central vein
- This zone is the most distal from arterial oxygen delivery (already relatively hypoxic)
- Zone III hepatocytes have the least glutathione reserve
With severe toxicity, necrosis can extend outward to involve entire lobules.
3. Factors That Increase Risk (Why Some People Are More Vulnerable)
| Risk Factor | Mechanism |
|---|---|
| Chronic alcohol use | Induces CYP2E1 (more NAPQI) and depletes baseline GSH |
| Fasting / malnutrition | Reduces GSH synthesis (requires cysteine, glycine, glutamate) |
| Anticonvulsants (phenytoin, phenobarbital, carbamazepine) | Induce CYP3A4 → more NAPQI |
| Isoniazid (INH) | Induces CYP2E1 → more NAPQI |
| Rifampicin, zidovudine | Induce oxidative CYP pathways |
| HIV/AIDS | Depleted GSH stores |
| Children <6 years | Relatively protected — greater hepatic sulfation capacity |
Chronic alcoholics are at higher risk, but there is still debate about whether acute alcohol co-ingestion is protective (competes for CYP2E1) or harmful (depletes GSH). — Harrison's Principles of Internal Medicine 22E
4. Four Clinical Stages of Toxicity
| Stage | Time | Features |
|---|---|---|
| Stage 1 | 0–24 h | Asymptomatic or nausea, vomiting, malaise. Liver enzymes normal. |
| Stage 2 | 24–72 h (days 2–3) | GI symptoms resolve. RUQ pain/tenderness appears. Transaminases rise. Oliguria may begin. |
| Stage 3 | 72–96 h (days 3–4) | Peak hepatotoxicity. Fulminant hepatic failure: jaundice, coagulopathy, encephalopathy, metabolic acidosis, renal failure (hepatorenal syndrome). ALT/AST may exceed 10,000 IU/L. |
| Stage 4 | Day 4 – 2 weeks | Recovery begins. Complete hepatic regeneration in survivors over 1–3 months. |
— Tintinalli's Emergency Medicine
5. Extrahepatic Injury
Because CYP450 enzymes (and prostaglandin H synthase) exist in organs outside the liver, toxicity is not exclusively hepatic:
- Renal tubular injury — via renal CYP2E1-mediated NAPQI generation in the proximal tubules
- Cardiac toxicity — rare; direct myocardial injury
- Pancreatitis — rare
- Massive ingestion (>500 mg/kg) — early-onset metabolic acidosis with elevated lactate even before liver failure, due to:
- GSH depletion → accumulation of 5-oxoproline (pyroglutamic acid)
- Direct NAPQI-mediated mitochondrial respiratory chain inhibition
6. Key Biochemical Markers
- Acetaminophen-protein adducts (APAP-Cys adducts) detectable in serum — serve as diagnostic biomarkers of hepatocellular damage
- AST/ALT > 10,000 IU/L with relatively low bilirubin is the biochemical signature of acetaminophen hepatotoxicity (hyperacute injury pattern)
- Blood levels >300 μg/mL at 4 hours post-ingestion predict severe liver damage
- Blood levels <150 μg/mL at 4 hours suggest hepatic injury is highly unlikely
7. Why the Antidote (N-Acetylcysteine) Works
NAC counters the pathophysiology at two levels:
- Early (within 8–12 h): Acts as a glutathione precursor (provides cysteine for GSH synthesis) and as a direct sulfur-containing substitute for GSH, directly conjugating NAPQI before it can bind hepatocyte proteins. Also enhances sulfation of the parent drug.
- Late (after hepatotoxicity is established): Acts as a free-radical scavenger, improves microcirculatory flow in the liver, and reduces the inflammatory cascade driven by necrotic hepatocyte damage.
Sources:
- Rosen's Emergency Medicine: Concepts and Clinical Practice, Chapter 138
- Tintinalli's Emergency Medicine: A Comprehensive Study, Chapter 190
- Harrison's Principles of Internal Medicine, 22nd ed., Chapter 351
- Lippincott Illustrated Reviews: Pharmacology
- Yamada's Textbook of Gastroenterology, 7th ed.
Generate ppt on this topic
Reading File
Reading File
Reading File
Running Command
Running Command
Writing File
Running Command
Editing File
Editing File
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
Create a quick reference PDF summarizing APAP toxicity stages & thresholds
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.