1. Classify plasma cell neoplasms. Discuss in detail the molecular pathogenesis, diagnosis, differential diagnosis, prognosis, cytogenetics, and complications of multiple myeloma. 2. Plasmacytoma short note 3. Minimal residual disease short note 4. Mast cell disorder short note
Reading File
Finding Sources
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have all the material needed. Let me compile the comprehensive answer.
1. Plasma Cell Neoplasms - Classification and Multiple Myeloma
Classification of Plasma Cell Neoplasms
Plasma cell neoplasms are B-cell proliferations in which neoplastic plasma cells virtually always secrete monoclonal immunoglobulin (M component) or immunoglobulin fragments. They account for approximately 15% of deaths from lymphoid neoplasms.
WHO Classification of Plasma Cell Neoplasms
| Category | Key Features |
|---|---|
| Multiple Myeloma (Plasma Cell Myeloma) | Most common; multiple lytic bone lesions, M protein in serum/urine |
| Smoldering (Asymptomatic) Myeloma | High serum M component; no CRAB symptoms |
| Solitary Plasmacytoma | Single osseous or extraosseous mass |
| Monoclonal Gammopathy of Undetermined Significance (MGUS) | Asymptomatic; M protein <3 g/dL; plasma cells <10% in marrow |
| Immunoglobulin-related (AL) Amyloidosis | Monoclonal light chains (usually λ) deposited as amyloid |
| Waldenström Macroglobulinemia | IgM hyperviscosity; associated with lymphoplasmacytic lymphoma |
| Heavy-Chain Disease | Rare; secretion of free heavy-chain fragments |
| Plasma Cell Leukemia | >20% circulating plasma cells; aggressive |
The M component types produced in myeloma (in order of frequency): IgG > IgA > IgD > IgM > light chain only > non-secretory.
Multiple Myeloma - Detailed Discussion
Epidemiology
Multiple myeloma causes approximately 1% of cancer deaths in Western countries. It is primarily a disease of older adults (median age ~65 years), with higher incidence in males and individuals of African descent. About 15,000 new cases are diagnosed annually in the United States.
Molecular Pathogenesis
Multiple myeloma arises from a postgerminal center B cell that has undergone somatic hypermutation and class switching. The pathogenesis follows a stepwise progression: Normal plasma cell → MGUS → Smoldering myeloma → Overt myeloma.
Key Molecular Events
1. Initiating Chromosomal Translocations (Primary Events)
The most important early genetic events involve translocations of the immunoglobulin heavy-chain (IgH) locus on chromosome 14q32 to oncogene loci. These juxtapose strong IgH enhancers with the partner gene, causing dysregulated expression:
| Translocation | Partner Gene | Frequency | Consequence |
|---|---|---|---|
| t(4;14)(p16;q32) | FGFR3, MMSET | ~15% | Poor prognosis |
| t(14;16)(q32;q23) | MAF | ~5% | Poor prognosis |
| t(11;14)(q13;q32) | Cyclin D1 (CCND1) | ~20% | Intermediate prognosis |
| t(6;14)(p21;q32) | Cyclin D3 | ~2% | - |
| t(14;20)(q32;q11) | MAFB | ~1% | Poor prognosis |
Cases without IgH translocations often show hyperdiploidy (trisomies of odd-numbered chromosomes 3, 5, 7, 9, 11, 15, 19, 21), which is associated with a better prognosis.
2. RAS Pathway Mutations (Secondary Events)
Activating mutations of N-RAS and K-RAS are present in ~40% of myelomas, driving autonomous proliferation via the MAPK/ERK pathway.
3. MYC Dysregulation
MYC overexpression occurs through translocations, amplifications, or secondary rearrangements. MYC drives cell cycle progression and is associated with progression from MGUS to overt myeloma.
4. Loss of Tumor Suppressors
- del(17p13) - Loss of TP53: ~10% of newly diagnosed, increases to 30-40% at relapse; worst prognosis
- del(13q14) - Loss of RB1 and/or miR-15a/miR-16-1: seen in ~50% of myelomas
- Loss of CDKN2C (p18INK4c) leads to uncontrolled cyclin D-CDK4/6 activity
5. NF-κB Pathway Activation
Mutations in TRAF3, TRAF2, CYLD, BIRC2/3, and NIK activate NF-κB in ~20% of myelomas, promoting plasma cell survival.
6. Epigenetic Changes
Mutations in histone-modifying enzymes (KDM6A/UTX, MMSET/NSD2 from t(4;14)) and DNA methyltransferases contribute to transcriptional reprogramming.
7. Bone Marrow Microenvironment
This is a critical driver of myeloma growth and drug resistance:
- Myeloma cells adhere to bone marrow stromal cells (BMSC) via VLA-4/VCAM-1 and syndecan-1 (CD138)
- Adhesion triggers IL-6 secretion by BMSCs → activates JAK/STAT3 pathway in myeloma cells (pro-survival, anti-apoptotic)
- RANKL (from myeloma cells and BMSCs) activates osteoclasts → bone destruction; simultaneously DKK-1 inhibits Wnt signaling → suppresses osteoblasts → net osteolysis
- VEGF promotes angiogenesis and tumor growth
- Immunosuppressive microenvironment: regulatory T cells, MDSCs, and PD-L1 expression facilitate immune evasion
Morphology
Bone Marrow:
- Plasma cells typically constitute >30% of marrow cellularity
- Cells have characteristic clock-face/cartwheel chromatin and perinuclear clearing (Golgi hof)
- Variants: Plasmablasts (vesicular chromatin, prominent nucleolus), Flame cells (fiery red cytoplasm), Mott cells (multiple grapelike cytoplasmic droplets containing Ig), Russell bodies (cytoplasmic Ig inclusions), Dutcher bodies (nuclear Ig inclusions)
- Multinucleated forms and bizarre cells may be seen
Bone Marrow Aspirate Histology:
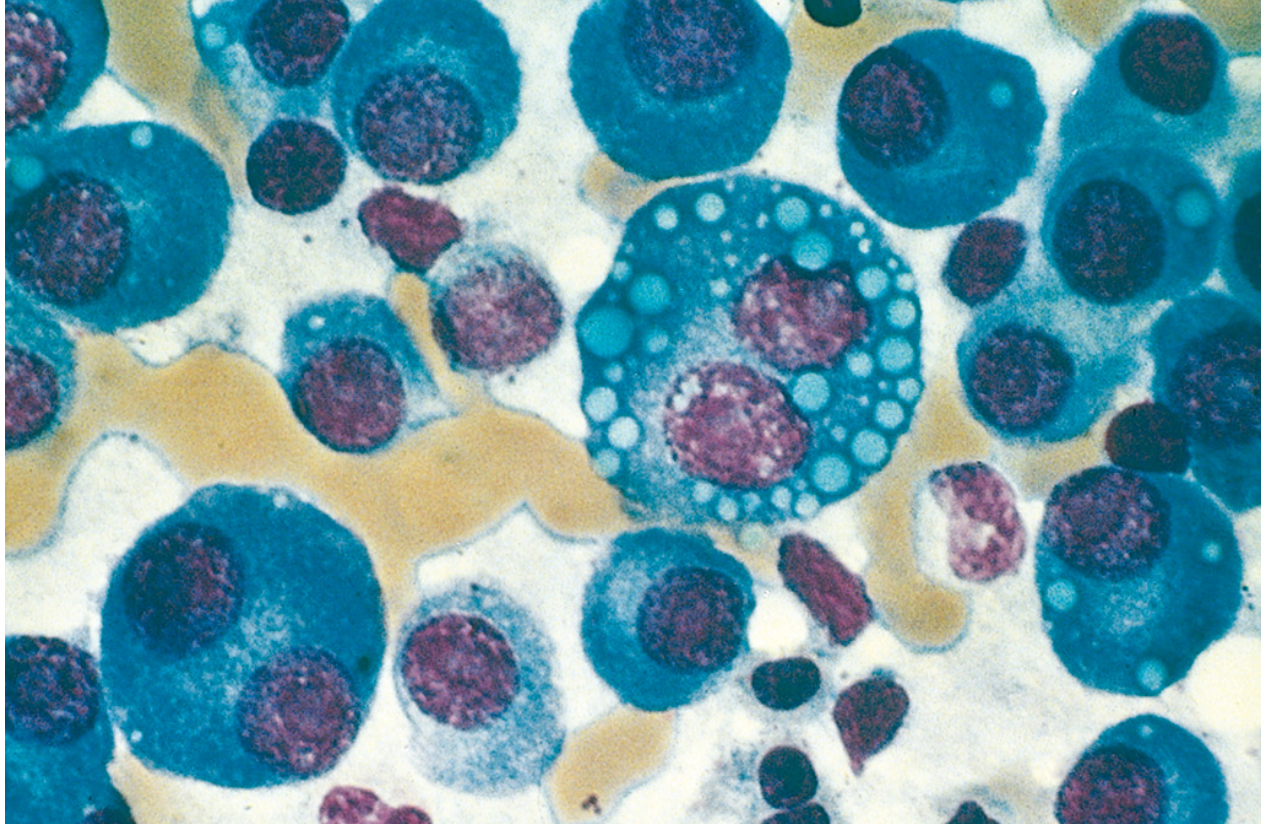 Fig: Multiple myeloma (bone marrow aspirate). Normal marrow cells are largely replaced by plasma cells including forms with multiple nuclei, prominent nucleoli, and cytoplasmic droplets containing immunoglobulin. (Robbins & Kumar Pathologic Basis of Disease)
Fig: Multiple myeloma (bone marrow aspirate). Normal marrow cells are largely replaced by plasma cells including forms with multiple nuclei, prominent nucleoli, and cytoplasmic droplets containing immunoglobulin. (Robbins & Kumar Pathologic Basis of Disease)
Peripheral Blood:
- Rouleaux formation - red cells stack in linear arrays due to elevated M protein (not specific; also seen in lupus, early HIV)
- Occasionally plasma cell leukemia when tumor cells flood peripheral blood
Immunophenotype:
- CD138+ (syndecan-1) - most specific marker
- CD38+ (strong)
- Often CD56+ (unlike normal plasma cells) - helpful in identifying small tumor populations
- Cytoplasmic κ or λ light chain (clonal restriction)
- CD19-, CD20- (distinguishes from normal plasma cells and lymphoplasmacytic lymphoma)
Diagnosis
Diagnostic Criteria (IMWG 2014):
A. Clonal bone marrow plasma cells ≥10% OR biopsy-proven plasmacytoma, PLUS any one of:
B. Myeloma-defining events (CRAB + SLiM):
- C - hyperCalcemia: serum calcium >0.25 mmol/L above upper normal or >2.75 mmol/L
- R - Renal insufficiency: creatinine >177 µmol/L or CrCl <40 mL/min
- A - Anemia: Hb <100 g/L or >20 g/L below lower normal
- B - Bone lesions: ≥1 osteolytic lesion on CT/PET-CT/skeletal survey
OR biomarkers of malignancy (SLiM):
- S - ≥60% clonal plasma cells in marrow
- Li - serum free Light chain ratio ≥100 (involved:uninvolved)
- M - >1 focal lesion on MRI (≥5 mm)
Laboratory Workup:
| Test | Finding in MM |
|---|---|
| Serum protein electrophoresis (SPEP) | M-spike (usually γ-region) |
| Immunofixation | Identifies Ig class and light chain type |
| 24-hour urine protein electrophoresis | Bence Jones proteinuria (free light chains) |
| Serum free light chains | Elevated κ or λ; skewed ratio |
| Complete blood count | Normocytic normochromic anemia |
| Serum calcium | Hypercalcemia |
| Creatinine/BUN | Renal impairment |
| Serum β2-microglobulin | Elevated; prognostic |
| LDH | Elevated in aggressive disease |
| Bone marrow biopsy | ≥10% clonal plasma cells |
| Skeletal survey/PET-CT | Lytic "punched-out" lesions |
Classic Radiographic Finding:
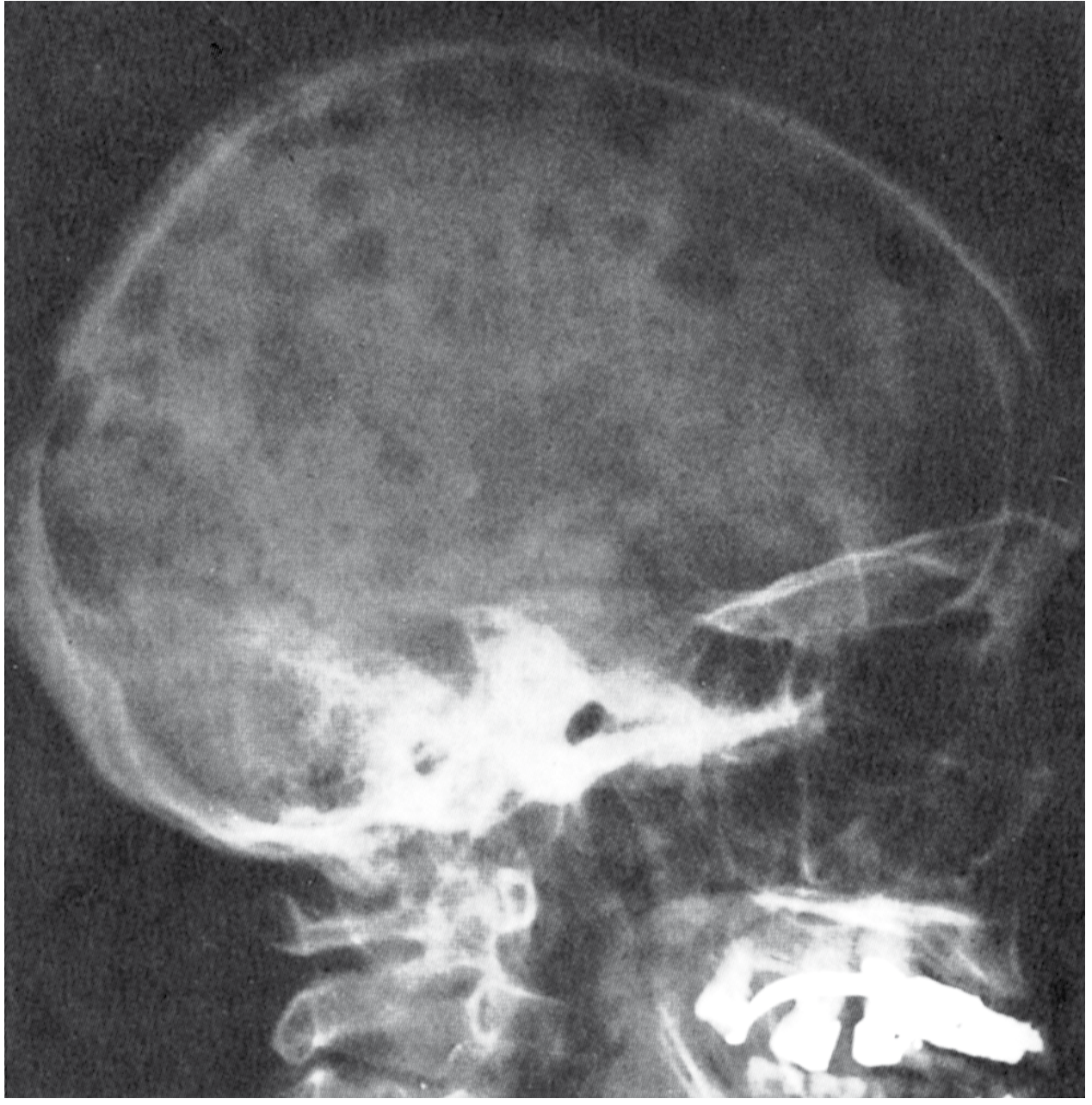 Fig: Multiple myeloma of the skull (radiograph, lateral view). The sharply punched-out bone lesions are most obvious in the calvaria. (Robbins & Kumar Pathologic Basis of Disease)
Fig: Multiple myeloma of the skull (radiograph, lateral view). The sharply punched-out bone lesions are most obvious in the calvaria. (Robbins & Kumar Pathologic Basis of Disease)
Differential Diagnosis
| Condition | Key Distinguishing Features |
|---|---|
| MGUS | Asymptomatic; M protein <3 g/dL; BM plasma cells <10%; no CRAB/SLiM criteria |
| Smoldering Myeloma | M protein ≥3 g/dL or BM plasma cells 10-60%; no CRAB criteria; no myeloma-defining biomarkers |
| Waldenström Macroglobulinemia | IgM paraprotein; hyperviscosity; no lytic bone lesions; MYD88 L265P mutation; lymphoplasmacytic lymphoma histology |
| Solitary Plasmacytoma | Single bone/soft tissue lesion; normal marrow elsewhere; no CRAB |
| AL Amyloidosis | Systemic amyloid deposits (Congo red positive); may have minor clonal plasma cells; no lytic lesions |
| Metastatic Carcinoma to Bone | Lytic lesions but with primary tumor; no M protein; no rouleaux |
| Reactive Plasmacytosis | <20% plasma cells; polyclonal; causes = infection, autoimmune |
| Lymphoplasmacytic Lymphoma | CD19+, CD20+; MYD88 mutation; IgM; no bone destruction |
| Heavy-Chain Disease | Only heavy chains secreted; no free light chains; rare |
Cytogenetics
Cytogenetic analysis (FISH on CD138-selected cells) is essential for risk stratification:
High-Risk Cytogenetics (Poor Prognosis):
- del(17p13) - TP53 loss - median OS ~24-26 months
- t(4;14)(p16;q32) - FGFR3/MMSET - median OS ~33 months
- t(14;16)(q32;q23) - MAF
- t(14;20) - MAFB
- Gain(1q21) - CKS1B amplification - adverse
- del(1p32) - CDKN2C loss
Standard-Risk Cytogenetics:
- Hyperdiploidy (trisomies of odd chromosomes) - best prognosis
- t(11;14)(q13;q32) - Cyclin D1 - standard risk (note: venetoclax-sensitive)
- del(13q) by itself - standard risk (when FISH-based)
ISS Staging + R-ISS (Revised International Staging System):
| Stage | Criteria | Median OS |
|---|---|---|
| R-ISS I | ISS I + standard-risk cytogenetics + normal LDH | 62 months |
| R-ISS II | Not I or III | 42 months |
| R-ISS III | ISS III + [high-risk cytogenetics OR high LDH] | 26 months |
ISS based on: β2-microglobulin and serum albumin.
- Stage I: β2M <3.5 mg/L + albumin ≥3.5 g/dL
- Stage II: Neither I nor III
- Stage III: β2M ≥5.5 mg/L
Prognosis
Favorable prognostic factors:
- Hyperdiploid karyotype
- t(11;14) - standard risk
- Low β2-microglobulin
- Low LDH
- Normal creatinine and calcium
- MRD negativity after treatment
- Age <65 years
- Good performance status
- Response to initial therapy
Adverse prognostic factors:
- del(17p), t(4;14), t(14;16), t(14;20), gain(1q21)
- High β2-microglobulin (>5.5 mg/L)
- Low albumin
- Elevated LDH
- Renal failure
- Plasmablastic morphology
- Plasma cell leukemia
- MRD positivity post-treatment
- High plasma cell proliferation index
Median survival with modern therapy (proteasome inhibitors + immunomodulatory drugs + anti-CD38 antibodies ± transplant): approximately 5-7 years for standard risk, 2-3 years for high risk.
Complications
The clinical features and complications arise from three main mechanisms: (1) plasma cell growth in tissues (especially bone), (2) production of abnormal immunoglobulins with pathologic physicochemical properties, and (3) suppression of normal humoral immunity.
1. Skeletal Complications
- Osteolytic bone lesions - the hallmark; due to uncoupled bone remodeling (increased osteoclast activity, suppressed osteoblast activity)
- Pathological fractures - vertebral compression fractures, long bone fractures
- Spinal cord compression - from vertebral collapse or epidural plasmacytoma
- Hypercalcemia - from osteoclast-mediated bone resorption → confusion, weakness, lethargy, constipation, polyuria; may precipitate renal failure
2. Renal Complications (Myeloma Kidney)
- Cast nephropathy (myeloma kidney) - the most common; Bence Jones proteins (free light chains) form casts in distal tubules and collecting ducts → tubular obstruction + giant cell reaction → tubulointerstitial nephritis
- Light chain deposition disease (LCDD) - non-fibrillar deposits of κ light chains in glomeruli and tubules → nephrotic syndrome
- AL amyloidosis - fibrillar λ light chain deposits; Congo red positive; glomerulonephritis/nephrotic syndrome
- Hypercalcemic nephropathy
- Hyperuricemic nephropathy
- Infections contributing to AKI
3. Hematologic Complications
- Anemia - normocytic normochromic; due to marrow replacement, renal EPO deficiency, cytokine suppression (IL-6, TNF-α)
- Leukopenia and thrombocytopenia - from marrow infiltration
- Hyperviscosity - particularly with IgA and IgM myelomas; causes visual disturbances, neurological symptoms, cardiac failure
4. Immunologic Complications
- Immunoparesis - suppression of normal (polyclonal) immunoglobulins → recurrent bacterial infections (especially encapsulated organisms: Streptococcus pneumoniae, Haemophilus influenzae)
- This is one of the leading causes of death in myeloma
5. Amyloidosis (AL)
- Primary AL amyloidosis in ~10% of myeloma patients
- Periorbital purpura ("raccoon eyes"), macroglossia, cardiomyopathy, hepatomegaly, peripheral neuropathy, nephrotic syndrome
6. Neurological Complications
- Hypercalcemia → encephalopathy
- Spinal cord/nerve root compression
- Peripheral neuropathy (from amyloid, myeloma infiltration, or treatment-related)
- Hyperviscosity → central neurological symptoms
- POEMS syndrome (rare variant): Polyneuropathy, Organomegaly, Endocrinopathy, M protein, Skin changes
7. Coagulation Abnormalities
- M proteins may interfere with coagulation factors, causing bleeding tendency
- Increased risk of thrombosis (especially with lenalidomide + dexamethasone)
2. Plasmacytoma - Short Note
A plasmacytoma is a localized neoplasm of clonal plasma cells arising at a single site, distinct from the diffuse skeletal involvement of multiple myeloma.
Types
A. Solitary Plasmacytoma of Bone (SPB)
- ~3-5% of plasma cell neoplasms
- Most commonly affects axial skeleton (vertebrae, pelvis, ribs, skull)
- Same age and sex distribution as myeloma (older males)
- Clinically: bone pain, pathological fracture; no CRAB features
- Serum/urine M protein may be mildly elevated in some
- Bone marrow elsewhere is normal (<5% plasma cells)
- Radiographically: solitary lytic lesion
- Histology: sheets of plasma cells replacing normal bone
- Natural history: virtually all SPB eventually progress to overt multiple myeloma, but this may take 10-20 years or longer
- Treatment: local radiation therapy; monitor for progression
B. Extramedullary (Extraosseous) Plasmacytoma
- Arises in soft tissues without involvement of bone or marrow
- Preferential sites: upper respiratory tract (nasal cavity, paranasal sinuses, nasopharynx, oropharynx) - accounts for ~80%
- Also: lungs, GI tract, thyroid, testes
- Older males predominantly
- M protein rarely elevated
- Prognosis is much more favorable than SPB: extraosseous plasmacytomas, particularly in the upper respiratory tract, are frequently cured by local resection or radiation
- Only ~15% progress to multiple myeloma (vs. nearly 100% for SPB)
Diagnosis
- Biopsy showing clonal plasma cells at single site
- Normal bone marrow biopsy (confirms it is not myeloma)
- No CRAB criteria
- Skeletal survey/PET-CT to exclude other lesions
- Serum and urine immunofixation, free light chains
Differential Diagnosis
- Multiple myeloma (multiple lesions, CRAB features, >10% marrow plasma cells)
- Metastatic carcinoma
- Giant cell tumor / Ewing sarcoma (in bone)
- Lymphoma (extraosseous sites)
- Reactive plasmacytosis
Source: Robbins & Kumar Pathologic Basis of Disease; Cummings Otolaryngology Head and Neck Surgery
3. Minimal Residual Disease (MRD) - Short Note
Definition
Minimal residual disease (MRD) refers to the detection of residual malignant cells that are below the threshold of detection by conventional light microscopy (i.e., <5% by morphology). It is the most sensitive measure of treatment response and one of the most powerful independent prognostic factors in hematologic malignancies.
Detection Methods
| Method | Sensitivity | Principle | Applications |
|---|---|---|---|
| Multiparameter Flow Cytometry (MFC) | 10⁻³ to 10⁻⁴ (0.01-0.001%) | Detects aberrant immunophenotypes unique to each patient's leukemia/myeloma | ALL, AML, MM |
| RQ-PCR (Real-time Quantitative PCR) | 10⁻⁴ to 10⁻⁵ | Detects patient-specific IgH/TCR gene rearrangements or fusion transcripts (BCR-ABL, MLL-AF4) | ALL, CML, CLL |
| Next-Generation Sequencing (NGS) | 10⁻⁵ to 10⁻⁶ | Deep sequencing of clonotypic sequences; identifies multiple leukemia-specific mutations | ALL, MM (clonoSEQ) |
| Digital Droplet PCR (ddPCR) | 10⁻⁵ to 10⁻⁶ | Partitions reaction into droplets; absolute quantification | AML, ALL, CML |
| ctDNA-based liquid biopsy | Variable | Detects circulating tumor DNA from blood plasma | Solid tumors, lymphoma |
Clinical Significance
In Acute Lymphoblastic Leukemia (ALL):
- MRD status after induction is the most important independent prognostic factor for disease-free and overall survival in both pediatric and adult ALL
- MRD-negative patients post-induction: disease-free survival ~70%
- MRD-positive patients: disease-free survival <40%
- MRD-positive patients should receive targeted therapy to reduce tumor burden, followed by allogeneic stem cell transplantation (SCT) if possible
- Phenotypic aberrations are unique to each patient and can be detected in up to 95% of individuals
- Bone marrow collection at diagnosis is essential for identifying individual markers
In Multiple Myeloma:
- Achieved via flow cytometry (EuroFlow panel) or NGS (clonoSEQ)
- MRD negativity is associated with significantly improved progression-free and overall survival
- Now considered a surrogate endpoint in clinical trials
In Solid Tumors (Emerging):
- ctDNA-based MRD after surgery shows high positive predictive value for relapse in colon, rectum, lung, bladder, and breast cancers
- Limitation: modest clinical sensitivity - substantial false-negative rate
- Coverage approved by CMS for colorectal cancer (Signatera MRD test, Natera)
- Active clinical trials testing MRD-guided adjuvant therapy
Technical Considerations
- Collection of bone marrow at diagnosis is essential to identify patient-specific markers
- 20-30% of adult ALL patients who are MRD-negative after induction still relapse (loss of sensitivity, clonal evolution, extramedullary disease)
- NGS and ddPCR offer superior sensitivity (parts-per-million range with multiplex bespoke assays)
- Liquid biopsies face challenges from biological background (clonal hematopoiesis) and technical noise
Source: Harrison's Principles of Internal Medicine 22E; Henry's Clinical Diagnosis and Management by Laboratory Methods
4. Mast Cell Disorders - Short Note
Definition
Mast cell disorders (mastocytosis) are clonal neoplastic proliferations of mast cells involving the skin, bone marrow, lymph nodes, spleen, liver, and/or other organs. The clinical spectrum ranges from benign self-limiting cutaneous disease to aggressive systemic illness.
WHO Classification
| Category | Subtypes | Key Features |
|---|---|---|
| Cutaneous Mastocytosis | Urticaria pigmentosa/MPCM, Diffuse cutaneous mastocytosis, Mastocytoma of skin | Skin only; extracutaneous organs not involved; common in children; often self-limited |
| Systemic Mastocytosis (SM) | Indolent SM (ISM), Smoldering SM (SSM), SM with AHN (SM-AHN), Aggressive SM (ASM), Mast cell leukemia (MCL) | Involvement of ≥1 extracutaneous organ ± skin |
| Mast Cell Sarcoma | - | Rare; destructive local tumor; high-grade |
Molecular Pathogenesis
- KIT D816V mutation is the hallmark: present in >90% of mastocytosis cases
- KIT (CD117) encodes a transmembrane receptor tyrosine kinase for stem cell factor (SCF)
- The D816V point mutation in exon 17 produces constitutive (ligand-independent) activation of KIT → growth factor-independent cell proliferation
- Important: KIT D816V is NOT specific to mastocytosis; also found in a subset of CBF-AML, GI stromal tumors, and melanomas
- Non-D816V KIT mutations also occur but are less common
- A subset with PDGFRA rearrangements also shows mast cell expansion but lacks KIT D816V - these cases are sensitive to imatinib
- SM frequently co-presents with other hematologic neoplasms (SM-AHN): chronic myelomonocytic leukemia (CMML) is most common, also MDS, MPN, AML
Clinical Features
From mediator release (histamine, prostaglandins, tryptase):
- Pruritus, urticaria, flushing, dermatographism
- Abdominal pain, diarrhea, nausea (GI histamine release)
- Anaphylaxis (often triggered by insect stings, NSAIDs, contrast media, alcohol)
- Hypotension, syncope
From organ infiltration:
- Bone pain, arthralgia, osteoporosis, pathological fractures
- Hepatomegaly, splenomegaly, lymphadenopathy
- Cytopenias from marrow infiltration
- Malabsorption
Skin (urticaria pigmentosa / MPCM):
- Tan/brown maculopapular lesions with urtication on stroking (Darier's sign)
- In children: often large, papular/bullous lesions; may spontaneously regress
Morphology
Skin:
- Aggregates of spindle-shaped mast cells in the papillary dermis, extending into reticular dermis
Bone Marrow (Systemic Mastocytosis):
- Multifocal, sharply demarcated compact aggregates of mast cells (key feature)
- Cells may be spindle-shaped and hypogranular on H&E
- Mixed infiltrates: mast cells + lymphocytes + eosinophils + histiocytes + fibroblasts, often with peripheral rim or central core of lymphocytes
- Associated fibrosis
- Tryptase-positive round cell infiltrates (TROCI) = compact infiltrates of hypergranular mast cells
- Reactive mast cells do NOT form compact aggregates (important distinction)
Immunophenotype / Markers
- Mast cell tryptase - most specific marker
- CD117 (KIT/c-kit) - strong expression
- CD25 and CD2 - aberrant expression (normal mast cells lack CD25/CD2) - used in diagnosis
- CD45, CD33, CD68 positive
- Myeloperoxidase negative
- Chloroacetate esterase positive
- Special stains: Giemsa, Toluidine blue (for granules in tissue)
Diagnostic Criteria for Systemic Mastocytosis
Major criterion:
- Multifocal, dense infiltrates of mast cells (≥15 mast cells in aggregates) in BM or other extracutaneous organs
Minor criteria (need 3 of 4 for SM diagnosis without major criterion, or 1 of 4 with major criterion):
-
25% of mast cells in BM biopsy are spindle-shaped or have atypical morphology
- Activating KIT D816V mutation
- Mast cells in BM express CD25 and/or CD2 (aberrant)
- Serum total tryptase persistently >20 ng/mL
Differential Diagnosis
- Reactive mast cell hyperplasia (no compact aggregates; seen in HCL, lymphoplasmacytic lymphoma, myeloid neoplasms)
- Basophilic leukemia (TROCI overlap)
- AML with abnormal mast cells
- Lymphoma with mast cell-rich stroma
Prognosis
- Cutaneous mastocytosis in children: excellent; often spontaneously resolves
- Indolent SM: near-normal life expectancy; main morbidity from mediator release
- SM-AHN: prognosis determined largely by the associated hematologic neoplasm
- Aggressive SM and Mast Cell Leukemia: median survival <1-2 years
Treatment Highlights
- Avoidance of triggers; H1/H2 antihistamines; mast cell stabilizers (cromolyn sodium)
- Epinephrine for anaphylaxis
- Bisphosphonates for osteoporosis
- Midostaurin (multi-kinase inhibitor including KIT): FDA-approved for advanced SM
- Avapritinib (selective KIT D816V inhibitor): approved for aggressive SM; highly effective against D816V mutation
- Imatinib: only effective in KIT D816V-negative cases (e.g., PDGFRA-rearranged)
- Cladribine, IFN-α: for cytoreduction in advanced SM
- Allogeneic SCT: for eligible patients with aggressive SM or MCL
Sources: Henry's Clinical Diagnosis and Management by Laboratory Methods; Harrison's Principles of Internal Medicine 22E; Robbins & Kumar Pathologic Basis of Disease
Summary Table: All Four Entities at a Glance
| Feature | Multiple Myeloma | Solitary Plasmacytoma | MRD | Mastocytosis |
|---|---|---|---|---|
| Cell of origin | Postgerminal-center plasma cell | Postgerminal-center plasma cell | N/A (measurement tool) | Mast cell (KIT-mutant progenitor) |
| Key mutation | IgH translocations, RAS, TP53, NF-κB | Same as MM (subset) | N/A | KIT D816V (>90%) |
| Marker | CD138, CD38, CD56 | CD138, CD38 | Clonotypic Ig/TCR, aberrant phenotype | Tryptase, CD117, CD25, CD2 |
| Main complication | CRAB (bone, renal, anemia, hypercalcemia) | Progression to MM (SPB ~100%) | Relapse if MRD+ | Anaphylaxis, bone disease, SM-AHN |
| Key imaging | Punched-out lytic lesions | Solitary lytic lesion | Not applicable | Skeletal sclerosis/lysis |
| Treatment | VRd/Dara-VRd + ASCT + maintenance | Radiation ± surgery | MRD-guided therapy escalation | Avapritinib (advanced), antihistamines |
This is a shared conversation. Sign in to Orris to start your own chat.