i have a presentation to present, what are the things that are missing? basics are not kept here, but i want to understand the basics in very detail, help me learn and understand that and explain each and every slide in detail and what to say in the images as well
ARMD Presentation - Complete Guide: What's Missing + Slide-by-Slide Explanation
WHAT IS MISSING FROM YOUR PRESENTATION
1. Basic Eye Anatomy (No Introductory Slide)
2. The Visual Cycle / Retinoid Cycle (Not Explained)
3. What Exactly Is the RPE? (Mentioned But Not Explained)
4. Bruch's Membrane - Its 5 Layers
5. What Is the Macula Clinically?
6. The Amsler Grid - How to Use It
7. How Anti-VEGF Injections Work Procedurally
8. No Slide on Normal Fundoscopy
THE BASICS YOU MUST KNOW COLD
THE EYE - Rapid Overview to Know Verbally
THE MACULA - What It Is
THE RPE - Why It's the Star of AMD
- Phagocytosis - eats 11-15% of shed photoreceptor outer segments every day (rods shed at dawn, cones throughout the day)
- Visual cycle - recycles vitamin A (retinal) to regenerate rhodopsin so you can keep seeing
- Outer blood-retinal barrier - tight junctions prevent leakage from choroid into retina
- Fluid transport - pumps fluid from subretinal space into choroid
- VEGF secretion (basolateral) - keeps the choriocapillaris healthy; in AMD this goes wrong
- Melanin - absorbs scattered light, reducing glare
- Vitamin A storage
BRUCH'S MEMBRANE - The 5 Layers (Must Know)
- RPE basement membrane
- Inner collagenous zone
- Elastic layer (middle)
- Outer collagenous zone
- Choriocapillaris basement membrane
THE VISUAL CYCLE (Why Lipofuscin Forms)
- Inhibits lysosomal enzymes (RPE can no longer digest shed outer segments)
- Activates complement (C3, MAC)
- Induces RPE apoptosis
- Fluoresces on fundus autofluorescence (FAF) imaging - this is why FAF is used to track AMD
SLIDE-BY-SLIDE WHAT TO SAY
Slide 1 - Title Slide
Slide 2 - Learning Objectives (BLANK - needs content)
- Describe the anatomy and physiology of the macula and RPE
- Explain the pathogenesis of dry and wet AMD including genetic and complement mechanisms
- Classify AMD using AREDS/Beckman criteria
- Apply a multimodal imaging approach
- Summarize management including AREDS2 supplements, anti-VEGF agents, and the 2023 FDA-approved GA treatments
Slide 3 - Definition & Overview
Slide 4 - Anatomy of the Macula
Slide 5 - Epidemiology & Prevalence
Slide 6 - Risk Factors: Non-Modifiable
Slide 7 - Risk Factors: Modifiable
Slide 8 - Genetics of AMD
Slide 9 - Pathogenesis Flowchart
Slide 10 - Complement Cascade in AMD
Slide 11 - Lipofuscin, A2E & RPE Toxicity
Slide 12 - VEGF Signalling Pathway
Slide 13 - Drusen (Introduction)
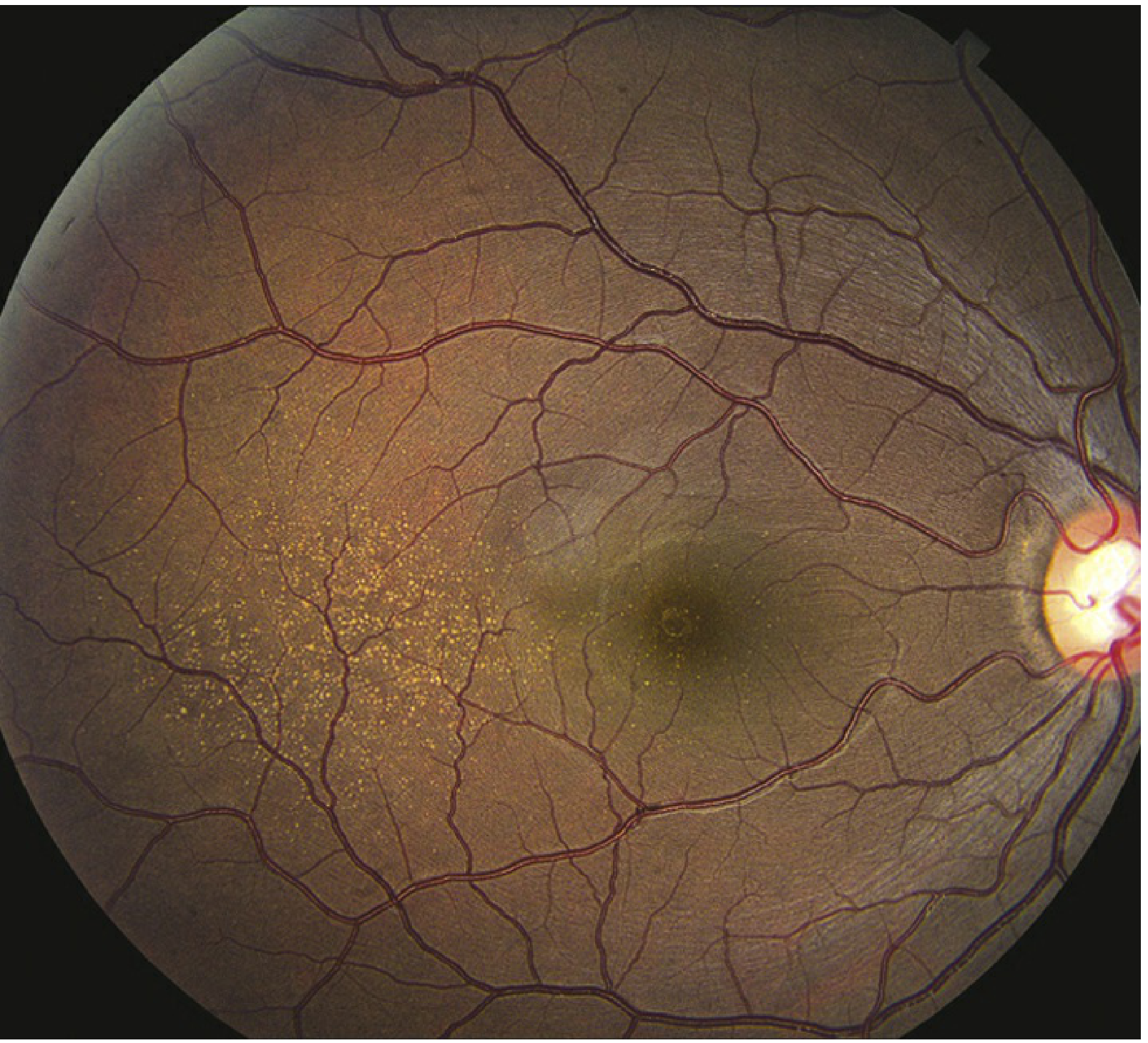
Slide 14 - Drusen Types & Clinical Significance
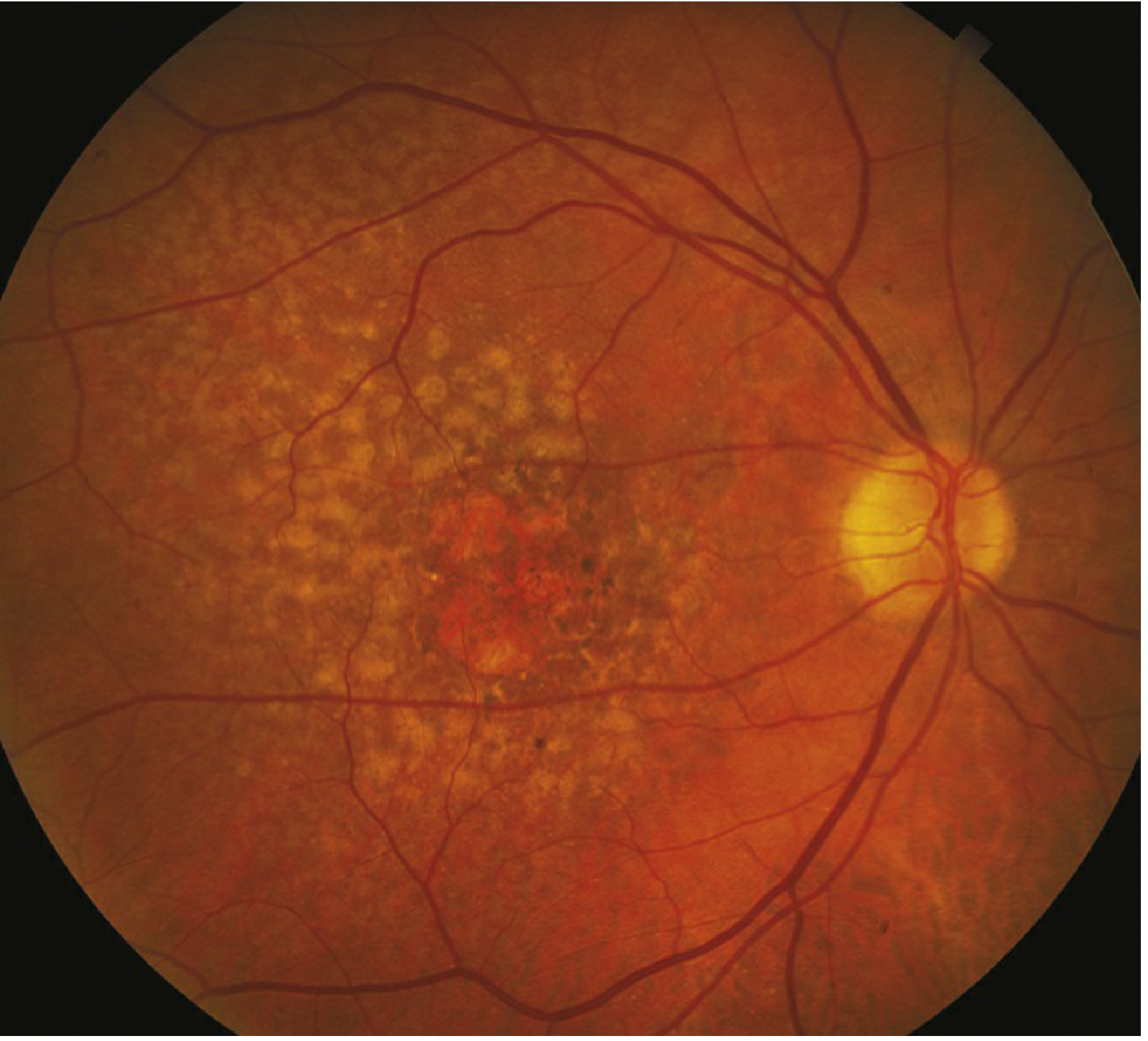
Slide 15 - Types of Macular Neovascularization (MNV)
Slide 16-17 - SDD vs Soft Drusen
Slide 18-19 - Polypoidal Choroidal Vasculopathy (PCV)
Slide 20-21 - Classification (AREDS/Beckman)
Slide 22 - Clinical Features: Dry AMD
Slide 23 - Clinical Features: Wet AMD
Slide 24-25 - Massive Subretinal Hemorrhage
Slide 26 - RPE Tear
Slide 27 - Disciform Scar
Slide 28 - Investigations & Imaging
Slide 29 - FAF Patterns in GA
Slide 30 - Dark Adaptation
Slides 31-32 - Advanced Functional Tests & Imaging Algorithm
Slide 33 - Differential Diagnosis
Slide 34-35 - AREDS Supplements
Slides 36-37 - Anti-VEGF Therapy
Slide 38 - Historical (Pre-Anti-VEGF) Treatments
Slides 39-40 - Dosing Strategies & Real-World Gap
Slides 41-42 - Treatment Flowchart & Complications
Slide 43 - Treatment Summary Table
Slides 44-45 - Tachyphylaxis & Combination Therapy
Slide 46 - Systemic Safety & Special Populations
Slides 47-50 - Clinical Trials (AREDS, MARINA, ANCHOR, CATT, VIEW, HAWK, YOSEMITE)
- MARINA (2006): Ranibizumab monthly → +7.2 letters vs -10.4 sham at 12 months. First trial showing vision GAIN.
- ANCHOR (2006): Ranibizumab vs PDT → +11.3 letters vs -9.5 PDT. Confirmed anti-VEGF superior to PDT.
- CATT (2011): Bevacizumab = ranibizumab monthly; PRN slightly less effective but non-inferior at 2 years.
- VIEW 1&2 (2012): Aflibercept q8w = monthly ranibizumab. Proven extended dosing.
- HAWK/HARRIER (2020): Brolucizumab non-inferior to aflibercept; 56% on q12w. Post-marketing vasculitis concern.
- YOSEMITE/LUCERNE (2022): Faricimab q16w non-inferior at 1 year; 53% maintained q16w at 2 years.
Slides 51-52 - Faricimab & Port Delivery System (PDS)
Slides 53-54 - Geographic Atrophy Treatments (2023 Historic Milestone)
Slides 55-56 - Gene Therapy & Emerging Treatments
Slide 57 - Biosimilars
Slide 58 - Angiopoietin-Tie Pathway
Slides 59-62 - AI, Recent Advances, Eylea HD
Slide 63 - Photobiomodulation (PBM)
Slides 64-65 - Stem Cell Therapy & Prognosis
Slides 66-68 - Prevention, Low Vision Rehab, Patient Education
Slide 69 - Clinical Approach Algorithm
Slide 70 - Key Take-Home Points
Slide 71 - References
LIKELY EXAMINER QUESTIONS - BE READY
-
"What is the visual cycle and how does it relate to AMD?" - Explain rhodopsin bleaching, all-trans retinal toxicity, RPE-mediated recycling, and A2E formation as the byproduct.
-
"Why does AMD cause central vision loss but not blindness?" - Macula is central 5.5mm; peripheral retina intact; patients can walk but can't read faces.
-
"What are the 5 layers of Bruch's membrane?" - RPE BM / Inner collagenous / Elastic / Outer collagenous / Choriocapillaris BM.
-
"Why do you not stop anticoagulation before intravitreal injection?" - Risk of systemic thromboembolism from stopping > risk of ocular hemorrhage; CATT data supports continuing.
-
"What is the difference between SRF, IRF, and PED on OCT?" - SRF: between photoreceptors and RPE (dark space); IRF: within retinal layers (cystoid spaces); PED: RPE lifted off Bruch's membrane.
-
"Why is ICGA better than FFA for PCV?" - ICG binds plasma proteins, stays in choroidal vessels longer, penetrates through RPE; FA cannot show the branching choroidal network or distinguish from occult CNV.
-
"Name the two FDA-approved GA treatments from 2023." - Pegcetacoplan (Syfovre) February 2023 - C3 inhibitor; Avacincaptad pegol (Izervay) August 2023 - C5 inhibitor.
-
"What is the mechanism of faricimab that makes it different?" - Bispecific antibody blocking both VEGF-A AND Angiopoietin-2; Ang-2 blockade restores Tie2-mediated vascular stability.
i have not added the epidemiology in nepal, help me add the data of nepal
age-related macular degeneration Nepal epidemiology prevalence
AMD ARMD epidemiology prevalence Nepal data statistics
AMD prevalence Nepal Bhaktapur retina study statistics data
macular degeneration Nepal Bhaktapur retina study prevalence
Nepal AMD blindness burden 2023 2024 global disease burden ARMD retinal disease
AMD Epidemiology in Nepal - Complete Data for Your Presentation
SLIDE CONTENT TO ADD
Suggested Slide Title: AMD in Nepal – Epidemiology & Burden
- Nepal has the highest ASPR for AMD-related low vision and blindness among 204 countries globally (GBD 2021)
- ASPR increased from 308.55 per 100,000 (1990) to 399.22 per 100,000 (2021); EAPC = +0.56
- AMD contributes to 8.7% of total blindness in Nepal (national population-based data)
- Retinal disease is the 2nd most common cause of blindness in Nepal; AMD is the leading cause within retinal disease
| AMD Type | Prevalence (95% CI) |
|---|---|
| Any AMD | 35.43% (33.25–37.65) |
| Mild Dry AMD | 26.02% (24.04–28.08) |
| Intermediate Dry AMD | 7.69% (6.52–8.99) |
| Geographic Atrophy | 1.02% (0.61–1.59) |
| Wet AMD | 0.70% (0.37–1.19) |
| Early AMD (overall) | 33.71% |
| Late AMD (overall) | 1.72% |
| Bilateral involvement | 63.13% of all AMD subjects |
- Among dry AMD patients: 36.53% had visual impairment; 2.78% were blind
- Among wet AMD patients: 46.1% had visual impairment; 23.08% were blind
- In the KMCTH hospital study (Pokharel et al., 2009): 52.5% of AMD patients had VA 6/24–6/60; 15% had VA <3/60
| Risk Factor | Nepal-Specific Finding |
|---|---|
| Smoking | Strongest significant risk factor (OR 1.02 per cigarette/day; P=0.007); 69.9% of hospital AMD patients had smoking history (Thapa 2011) |
| Pseudophakia (post-cataract surgery) | OR 1.45 (95% CI: 1.12–1.87; P=0.005) — unique to Nepal; prolonged UV exposure during surgery or pre-op AMD |
| Hypertension | 45.4% of AMD patients had hypertension (KUMJ 2011); OR 1.21 in multivariate (Bhaktapur Retina Study) |
| Agriculture occupation | 42.5% of AMD patients; prolonged outdoor sunlight/UV exposure |
| Illiteracy | 75.6% of AMD patients illiterate; barrier to early diagnosis |
| Age | Prevalence 19.3% at >71 years (KMCTH study) |
- Mean age at presentation: 69.5 years
- Male predominance: 58.9% male (contrast with global female predominance)
- Dry AMD: 62.4% | Wet AMD: 37.6%
- Bilateral involvement: 88.7% of patients
- Presenting VA <6/18 in 71.4% of AMD eyes — reflects late presentation
- Most common pattern: One eye dry + fellow eye wet = 44% of patients
- Only 7.6% of the Bhaktapur population was aware of AMD
- Only 7.5% knew that smoking worsens AMD
- Only 7.4% knew sunlight exposure is a risk
- Awareness was lower in: older age groups, females, illiterates, farmers, and those with poor visual acuity — the exact groups most at risk
- Prevalence of any retinal disorder: 52.37% in those ≥60 years
- AMD was the most common retinal disease (35.43%)
- Followed by: hypertensive retinopathy (4.35%), ERM (3.66%), BRVO (2.90%), diabetic retinopathy (2.15%)
KEY MESSAGES FOR VERBAL PRESENTATION
WHERE TO INSERT IN YOUR PRESENTATION
REFERENCES TO CITE ON THE SLIDE
- Thapa R et al. Prevalence of and risk factors for AMD in Nepal: the Bhaktapur Retina Study. Clin Ophthalmol 2017; PMID 28579747
- Thapa R et al. Prevalence, Pattern and Risk Factors of Retinal Diseases in Nepal: the Bhaktapur Retina Study. Clin Ophthalmol 2020; PMID 32801619
- Thapa R et al. Age-related macular degeneration in Nepal. Kathmandu Univ Med J 2011; PMID 22609500
- Thapa R et al. Population awareness of AMD in Nepal. BMC Ophthalmol 2015; PMID 26714483
- Pokharel S et al. Pattern of AMD. JNMA 2009; PMID 20795460
- BMC Public Health 2024 - Global burden of AMD 1990–2021 (GBD 2021 data - Nepal highest ASPR globally)
i have data of our hospital BEH from jan 2025-dec 2025, total opd patient: 1791779, AMD: 1402, male: 812, female: 590
BEH Hospital Data - Calculated Statistics
Raw Numbers
| Parameter | Value |
|---|---|
| Total OPD (Jan–Dec 2025) | 1,791,779 |
| Total AMD patients | 1,402 |
| AMD as % of total OPD | 0.08% (0.78 per 1,000 OPD visits) |
| Monthly average | ~117 AMD cases/month |
Sex Distribution
| n | % | |
|---|---|---|
| Male | 812 | 57.9% |
| Female | 590 | 42.1% |
| M:F ratio | 1.38 : 1 |
Suggested Slide Content (add to your Nepal Epidemiology slide)
"At our institution, 1,402 AMD cases were recorded over 12 months out of 1,791,779 total OPD attendees — a prevalence of 0.08% among all OPD patients (~0.78 per 1,000 visits). Monthly average: ~117 new AMD cases per month."
Male predominance: 57.9% male vs 42.1% female (M:F = 1.38:1) This is consistent with the Tilganga hospital data (Thapa et al., 2011: 58.9% male) and stands in contrast to the global trend of slight female predominance — possibly reflecting healthcare-seeking behaviour patterns in Nepal where males access tertiary care more readily.
Key Talking Points for Presentation
How to Frame the Full Nepal Slide (Combined)
i am not able to understand anything, explain me from the very basics
PART 1 — THE EYE: How It Works
Step 1: Think of the Eye Like a Camera
| Camera part | Eye equivalent |
|---|---|
| Camera body (outer shell) | Sclera (white wall) |
| Lens | Cornea + Lens (focuses light) |
| Dark interior | Vitreous (gel filling the eye) |
| Film / sensor | Retina (catches the image) |
Step 2: The Eye in Cross-Section
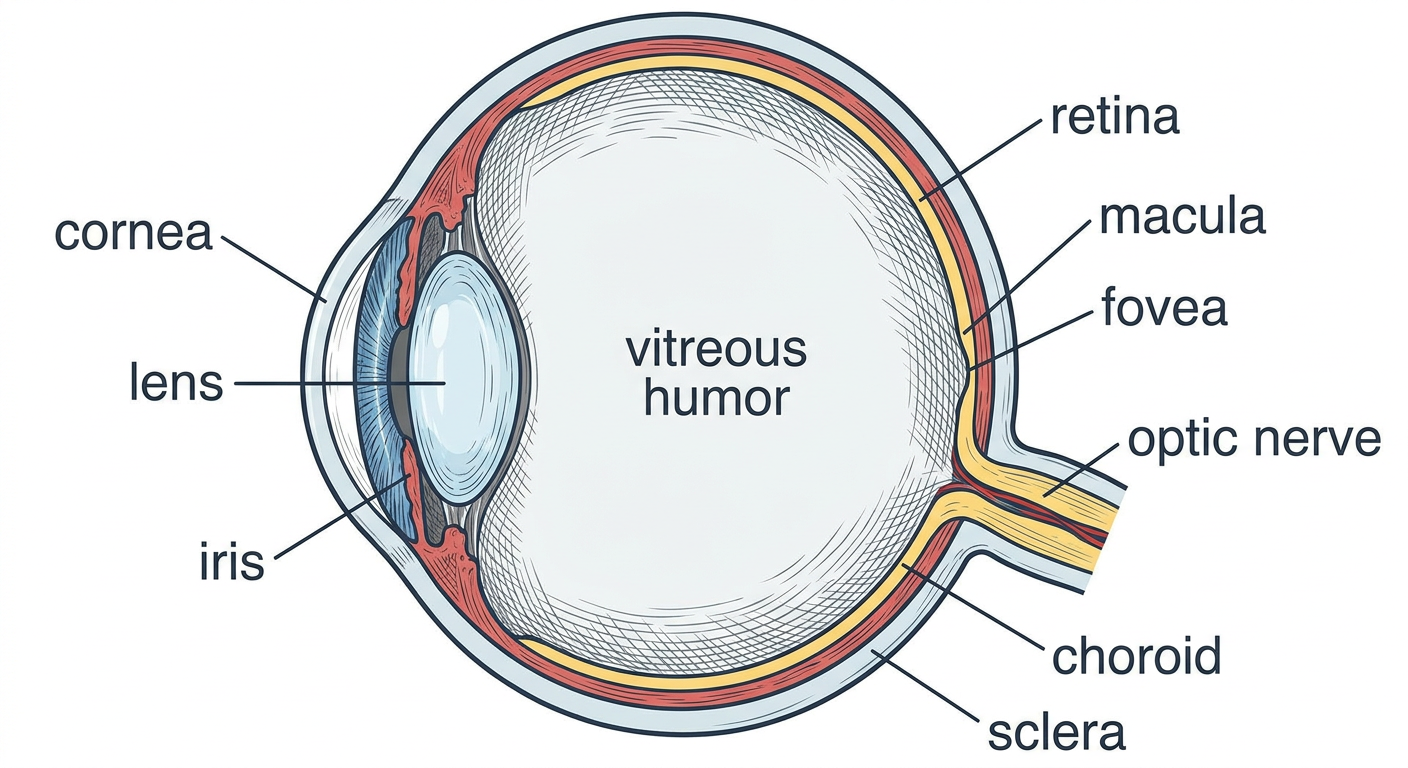
- Cornea - the clear window at the front. Does 70% of the focusing. No blood vessels (gets oxygen from air directly).
- Iris - the colored ring. Controls how much light enters by making the pupil bigger or smaller.
- Lens - sits just behind the iris. Does the fine-tuning of focus (especially for near objects). This is what goes cloudy in cataract.
- Vitreous - the jelly filling the middle. Not important in AMD.
- Retina - the thin film lining the back wall. This is the "sensor." It receives light and converts it to electrical signals.
- Macula - a special zone in the CENTER of the retina. This is the VIP area of vision.
- Fovea - the very center dot of the macula. Highest precision vision happens here.
- Optic nerve - the cable that carries signals from the retina to the brain.
- Choroid - a layer of blood vessels BEHIND the retina. It feeds the retina with blood and nutrients.
- Sclera - the tough white outer coat.
Step 3: The Macula — Why It Matters So Much
- The whole dartboard = retina (covers everything)
- The bullseye in the center = macula (the best part, only 5.5 mm)
- The very center dot = fovea (sharpest point, only 1.5 mm)
- Patient cannot read (macula is damaged)
- Patient can still walk around without falling (peripheral retina is fine)
- Patient is legally blind but NOT totally blind - they still have peripheral vision
PART 2 — THE RETINA: Layers You Must Know
Step 4: The Retinal Layers
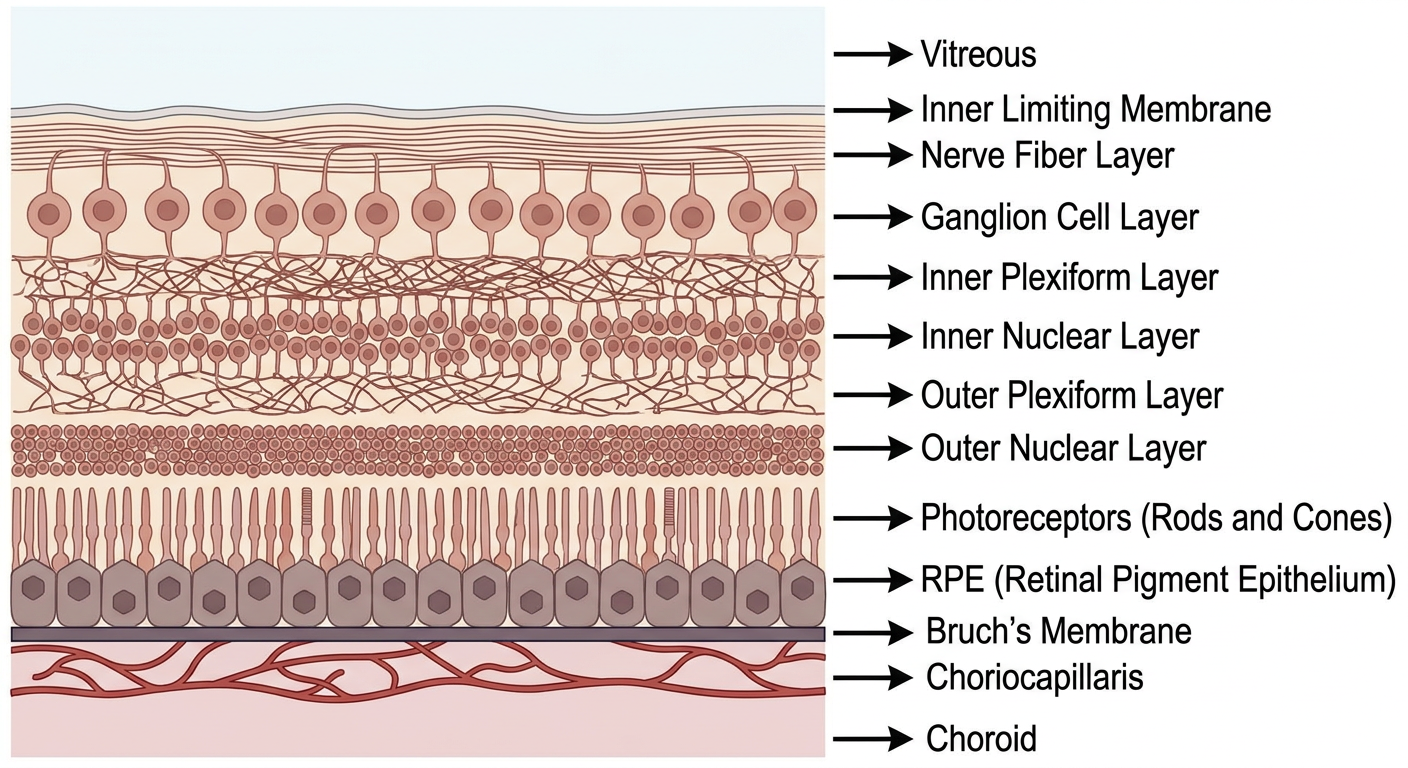
- Inner Limiting Membrane - just a physical boundary
- Nerve Fiber Layer - axons of ganglion cells (the "wires")
- Ganglion Cell Layer - nerve cells that send signals to brain
- Inner Plexiform Layer - connections/synapses
- Inner Nuclear Layer - bipolar cells, amacrine cells
- Outer Plexiform Layer - connections/synapses
- Outer Nuclear Layer - contains the cell bodies (nuclei) of rods and cones
- Photoreceptors (Rods & Cones) - THE MOST IMPORTANT. These cells catch light.
- RPE (Retinal Pigment Epithelium) - a single layer of cells. THE STAR OF AMD. More below.
- Bruch's Membrane - a thin sheet between RPE and choroid. THE FILTER.
Step 5: Rods vs Cones — The Two Types of Photoreceptors
| Rods | Cones | |
|---|---|---|
| Number | ~120 million | ~6 million |
| Location | All over retina EXCEPT fovea | Mainly concentrated in fovea |
| What they see | Low light, movement, peripheral vision | Color, fine detail, bright light |
| Used for | Night vision, navigation | Reading, faces, TV |
| Pigment used | Rhodopsin | Cone-specific opsins (R, G, B) |
PART 3 — THE RPE: The Most Important Cell in AMD
Step 6: What Is the RPE?
- They clean up the mess (phagocytosis)
- They control what comes in and out (barrier function)
- They maintain the lighting (visual cycle)
- They keep the plumbing working (fluid transport)
- If the security guards are fired (RPE dies) → the building collapses (photoreceptors die) → blindness
Job 1: PHAGOCYTOSIS (Cleaning)
Job 2: VISUAL CYCLE (Recycling visual pigment)
- Light hits rhodopsin in rods → rhodopsin bleaches → 11-cis retinal becomes all-trans retinal
- All-trans retinal is toxic to the photoreceptor
- The RPE collects it, converts it back to 11-cis retinal
- Returns it to the photoreceptor to make new rhodopsin
- You can see again
Job 3: OUTER BLOOD-RETINAL BARRIER
Job 4: FLUID TRANSPORT
Job 5: VEGF SECRETION
Step 7: Bruch's Membrane — The Filter
- RPE basement membrane
- Inner collagenous zone
- Elastic layer (middle - most important)
- Outer collagenous zone
- Choriocapillaris basement membrane
PART 4 — NOW, WHAT IS AMD?
Step 8: AMD in Plain Language
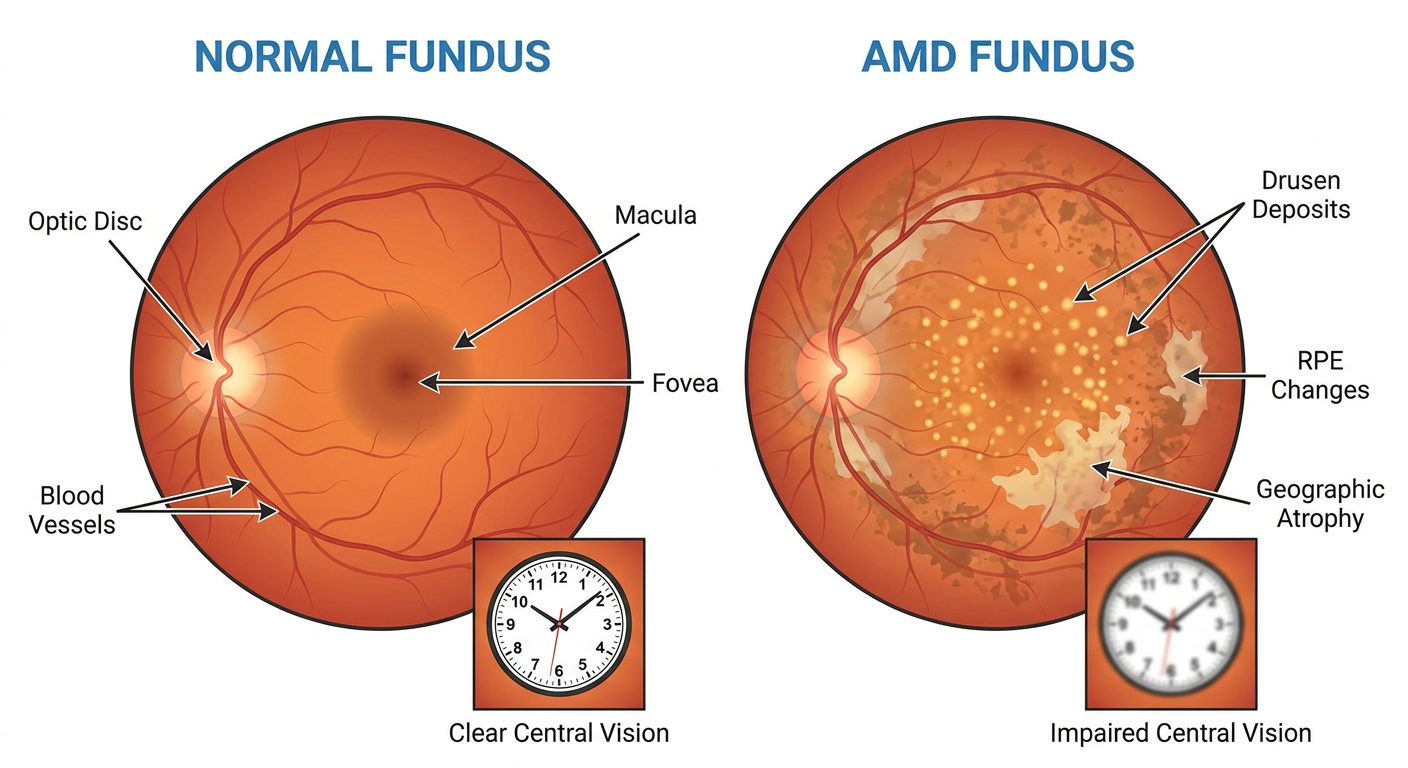
- On the left: a normal fundus. Clean, orange-red background. Clear macula. Healthy vessels. Foveal reflex visible at the center.
- On the right: AMD fundus. Yellow dots (drusen). Pale washed-out areas (RPE changes). Bare patches (geographic atrophy). The clock in the center shows blurry, unclear vision.
Old age + Smoking + Bad genes
↓
RPE gets stressed → cannot clean properly
↓
Debris piles up in Bruch's membrane
↓
DRUSEN form (yellow deposits you see on fundoscopy)
↓
┌──────────────────┬──────────────────┐
▼ ▼
RPE cells slowly die RPE becomes hypoxic
→ geographic atrophy → too much VEGF released
→ DRY AMD (late) → new blood vessels grow
→ bleed and leak
→ WET AMD (late)
Step 9: Dry vs Wet AMD — The Core Distinction
| Dry AMD | Wet AMD | |
|---|---|---|
| Other names | Non-neovascular, Atrophic | Neovascular, Exudative |
| What happens | RPE slowly dies → geographic atrophy | New blood vessels grow through Bruch's membrane |
| Speed | Slow - months to years | Fast - days to weeks |
| Vision loss | Gradual, insidious | Sudden, rapid |
| Symptom | Slowly worsening blur, dark adaptation difficulty | Sudden distortion (metamorphopsia), central blur |
| How common | 85-90% of all AMD cases | 10-15% of cases |
| Causes severe loss | 10% of severe vision loss | 90% of severe vision loss |
| Treatment | AREDS2 supplements (slow it down) / new: complement inhibitors | Anti-VEGF injections (gold standard) |
Step 10: What the Patient Actually Feels
- "I can't read the newspaper anymore"
- "My central vision is blurry but I can still walk around"
- "It has been slowly getting worse for years"
- On Amsler grid: lines appear slightly wavy or missing
- "Overnight, the center of my vision disappeared"
- "The door frame looked wavy when I woke up this morning" ← This is metamorphopsia
- "There is a dark spot in the middle of my vision" ← central scotoma
- Urgent — needs treatment within days
Step 11: The Amsler Grid — Simple But Important
Step 12: Drusen — What Are They Exactly?
- Hard drusen - small (<63 µm), sharp edges, relatively safe
- Soft drusen - large (>125 µm), fuzzy edges, HIGH RISK. When multiple soft drusen merge together (confluent), risk of progressing to late AMD is very high.
Step 13: The Three Questions Examiners Always Ask
- SRF (Subretinal Fluid): Fluid between the photoreceptors and the RPE. A dark (hyporeflective) space on OCT between these two layers. Comes from leaking new vessels.
- IRF (Intraretinal Fluid): Fluid within the retinal layers themselves, forming cystoid (round, dark) spaces within the retina. Worse prognosis than SRF.
- PED (Pigment Epithelial Detachment): The RPE itself gets lifted off Bruch's membrane by fluid or fibrovascular tissue underneath. On OCT it looks like a dome or bump of elevated RPE.
YOUR LEARNING ROADMAP
✅ DONE NOW:
Eye anatomy → Retinal layers → RPE functions
Bruch's membrane → Drusen → Dry AMD → Wet AMD
NEXT STEP: (tell me when ready)
Pathogenesis in detail
(Complement cascade, VEGF pathway,
exactly how AMD progresses step by step)
THEN:
Clinical presentation + Investigations
(OCT interpretation, FFA, FAF)
THEN:
Treatment
(AREDS2, anti-VEGF drugs, GA treatments)
Am I clear on the eye anatomy and layers so far?
1. central macula is affected, peripheral is normal but why? 2. RPE, photoreceptor, ONL 3. RPE pump failuure? 4. Wet because wet occurs only in ~10% but affecting 90 % of the affected. 5. metamorphopsia: fluid accumulates beneath photoreceptor causing it to lift and shift the photoreceptor cell and brain percieves it as wavy
Your Score: 4/5 solid, 1 needs a small correction
The three key structures in AMD = RPE + Bruch's membrane + Choriocapillaris Drusen form because of phagocytosis failure, not pump failure
Ready for the Next Level?
- What exactly goes wrong inside the RPE cell (lipofuscin, A2E)
- Why the complement system attacks the retina
- How VEGF drives new vessel growth in wet AMD
- Why smoking makes it worse
next
next
PART 5 — PATHOGENESIS: Why Does AMD Actually Happen?
CHAPTER 1: The RPE Gets Overworked and Starts Accumulating Garbage
- Eats shed outer segments (phagocytosis)
- Runs the visual cycle (recycles vitamin A)
- Pumps waste through Bruch's membrane into the blood
- Cigarette smoke (oxidative damage)
- UV light (photo-oxidative damage — especially in Nepal where outdoor farming is common)
- Bad genes (CFH mutation — more on this shortly)
- High blood pressure (damages choroidal blood supply)
Inside the RPE cell — The Visual Cycle Goes Wrong
Light hits rhodopsin
↓
11-cis retinal → all-trans retinal (toxic)
↓
RPE collects it → converts back to 11-cis retinal
↓
Returns to photoreceptor → rhodopsin regenerated ✅
- Poisons the lysosome (the RPE's own waste-disposal system shuts down)
- RPE can no longer properly digest shed outer segments
- Triggers the complement system (immune attack — Chapter 2)
- Eventually causes the RPE cell to self-destruct (apoptosis)
Outside the RPE — Bruch's Membrane Gets Clogged
CHAPTER 2: The Complement System Gets Activated (The Immune Attack)
What is the complement system?
- C3a is released → attracts macrophages and causes inflammation
- C3b sticks to surfaces → marks them for destruction
- C3b leads to splitting of C5
- C5 splitting produces C5a (more inflammation) and MAC (membrane attack complex, C5b-9)
- The MAC punches holes in cell membranes → cell dies
The Policeman: CFH (Complement Factor H)
The AMD Genetic Problem: CFH Y402H Mutation
Normal person:
Drusen accumulate → complement tries to activate → CFH stops it → controlled ✅
AMD patient (mutant CFH):
Drusen accumulate → complement activates → CFH CANNOT stop it
↓
C3 → C3a + C3b → C5 → C5a + MAC
↓
MAC deposits on RPE cells and choriocapillaris
↓
RPE cells are punched full of holes → they die
↓
GEOGRAPHIC ATROPHY (bare patches where RPE used to be)
Why Does Smoking Make This Worse?
- Directly oxidizes RPE cells and Bruch's membrane → accelerates A2E accumulation and Bruch's thickening
- Reduces CFH activity → less protection against complement activation
CHAPTER 3: Two Roads From Drusen — Dry or Wet?
Road 1 → DRY AMD (Geographic Atrophy)
- The photoreceptors above it lose their support system → they die too
- The choriocapillaris below it also atrophies (no RPE to send signals to maintain it)
- A bare, pale, well-defined patch appears on fundoscopy → Geographic Atrophy
Road 2 → WET AMD (Neovascular AMD)
Hypoxic RPE → HIF-1α activated → VEGF-A produced
↓
VEGF-A binds VEGFR-2 receptor on nearby endothelial cells
↓
Endothelial cells get the signal: "Grow! Divide! Make new vessels!"
↓
New blood vessels (choroidal neovascularization = CNV) sprout
from the choriocapillaris
↓
They try to grow toward the oxygen-starved area
↓
They BREACH THROUGH BRUCH'S MEMBRANE
↓
They enter the subretinal space (between RPE and photoreceptors)
↓
These vessels are ABNORMAL — thin-walled, fragile, leaky
↓
They leak fluid → subretinal fluid, intraretinal fluid
They bleed → subretinal hemorrhage
↓
Photoreceptors drown in fluid and blood → rapid vision loss
CHAPTER 4: Why Genetics Matter — The CFH + ARMS2 Story
Gene 1: CFH (Chromosome 1q32)
Gene 2: ARMS2/HTRA1 (Chromosome 10q26)
Genetic susceptibility × Environmental exposure = AMD phenotype
- CFH mutation alone → not certain AMD
- Smoking alone → increased risk but not certain
- CFH mutation + ARMS2 + smoking + age → very high risk
THE FULL PATHOGENESIS IN ONE FLOW
AGING + SMOKING + BAD GENES (CFH, ARMS2)
↓
RPE stressed → visual cycle produces A2E (toxic waste)
A2E poisons RPE lysosomes → phagocytosis fails
↓
Undigested waste + lipids pile up in Bruch's membrane
→ DRUSEN form
↓
Drusen activate COMPLEMENT SYSTEM
Mutant CFH cannot stop it
C3 → C3a + C3b → C5 → MAC
MAC attacks RPE cells
↓
┌───────────────────────┐
↓ ↓
RPE dies slowly RPE becomes hypoxic
Complement attack HIF-1α activated
Geographic Atrophy VEGF-A overproduced
DRY AMD (late) New vessels breach Bruch's
Leak + bleed in subretinal space
WET AMD (late)
Quick Check — 3 Questions
1. VEGF-A; VEGF-A binds to VEGFR-B in the nearby endothelium--new vessel formation from the choriocapillaries through the bruch's membrane into the RPE--leak--intraretinal fluid, subretinal fluid and bleed- subretinal hemorrhage. 2. CFH mutation doesn't bind to c3b and causes activation of c5 leading to c5a and c5b-9(MAC) activation--drusen formation? 3. visual cycle, even though visual cycle remains normal, 2 all-trans-retinal binds inside RPE lysosome with fatty acid leading to formation of phosphatidylethanolamine. all trans retinal and phosphatidylethanolamine are toxic to RPE cells leading to defective phagocytosis and accumulation of lipofusin(A2E bis-retinoid)
Drusen form FIRST (from phagocytosis failure + Bruch's clogging)
↓
Drusen THEN activate complement
↓
CFH mutation means complement cannot be switched off
↓
MAC attacks RPE → Geographic Atrophy
- The visual cycle running imperfectly
- Two molecules of all-trans retinal involved
- Condensation with phosphatidylethanolamine inside the RPE lysosome
- The result being A2E — a bis-retinoid
- Consequence: defective phagocytosis, lipofuscin accumulation
Your Score: 3/3 with minor refinements
✅ VEGFR-2 (not VEGFR-B) ✅ Drusen come FIRST → then activate complement (not the other way around)
Ready for Part 3?
- ✅ Anatomy and layers
- ✅ What the RPE does
- ✅ How AMD develops (A2E → drusen → complement → GA / VEGF → wet AMD)
next
PART 3 — CLINICAL FEATURES: What the Patient Tells You and What You See
THE GOLDEN RULE FIRST
AMD = Central vision loss. Peripheral vision preserved. Never total blindness.
SECTION A: WHAT THE PATIENT TELLS YOU (Symptoms)
Dry AMD Patient — The Slow Decline
- "Reading has become difficult — I need brighter light"
- "I struggle to see in dim light / entering a dark room" ← this is dark adaptation delay — the EARLIEST symptom, before anything is visible on fundoscopy
- "Colors look washed out"
- Often — nothing at all. Patient is asymptomatic.
- "There is a blank/dark patch in the center of my vision" ← central scotoma
- "I can see around things but not the center"
- "I can walk fine but I cannot recognize your face"
- "I cannot read even with glasses"
Wet AMD Patient — The Emergency
- "Doctor, when I woke up this morning, the door frame looked wavy" ← metamorphopsia — THE classic symptom
- "Lines on the floor tiles are not straight anymore"
- "There is a dark/gray spot in the center of my vision" ← central scotoma
- "My vision suddenly blurred in one eye"
- "I was reading and the words suddenly disappeared"
SECTION B: WHAT YOU SEE (Signs on Examination)
Step 1: Visual Acuity
- Early AMD: VA may be normal (6/6) — drusen alone may not affect acuity
- Intermediate AMD: mild reduction, often 6/9 to 6/12
- Late dry (GA): moderate to severe reduction, 6/18 to 6/60 or worse when fovea is involved
- Wet AMD: can drop rapidly from 6/6 to 6/60 within weeks if untreated
Step 2: Amsler Grid Testing
- Patient wears their reading glasses if they use them
- Cover one eye
- Hold grid at 30 cm (arm's length)
- Ask: "Can you see the central dot?"
- Ask: "Are all the lines straight?"
- Ask: "Are there any missing, blurry, or distorted areas?"
- Lines appear wavy/bent → metamorphopsia (wet AMD, fluid displacing photoreceptors)
- Lines or areas missing → scotoma (RPE/photoreceptor loss in GA)
- Central dot cannot be seen → advanced foveal involvement
Step 3: Dilated Fundoscopy — What You Actually See
- Optic disc — pale yellowish-orange circle, slightly nasal
- Macula — temporal to the disc, darker reddish-brown area
- Foveal reflex — tiny bright dot at the very center of the macula (from the concavity of the fovea)
- Blood vessels — radiating from the disc
SIGNS IN DRY AMD:
| Hard drusen | Soft drusen |
|---|---|
| Small, <63 µm | Large, >125 µm |
| Sharp, distinct edges | Fuzzy, indistinct edges |
| Scattered | May be confluent (merging) |
| Low risk | HIGH RISK |
| Look like white dots | Look like pale blobs |
- Hyperpigmentation — dark clumps. This is RPE cells bunching together or migrating in response to stress. Appears as dark pigment deposits around/over drusen.
- Hypopigmentation — pale areas. This is thinning and atrophy of RPE. Looks washed out.
- Well-defined, scalloped edges
- Typically starts AROUND the fovea (pericentral pattern) — sparing central vision initially
- Slowly enlarges concentrically
- When it finally swallows the fovea → severe central vision loss
- Both eyes usually affected — often asymmetrically
SIGNS IN WET AMD:
SECTION C: THE FELLOW EYE RULE
- Always examine the fellow eye carefully
- Look for soft drusen or RPE changes in the fellow eye (signs of intermediate AMD)
- Teach the patient Amsler grid for BOTH eyes — check each eye separately every day
- Tell them: "If the other eye develops any distortion, come immediately"
SECTION D: HOW TO EXAMINE AN AMD PATIENT — The Sequence
1. HISTORY
- Age, onset, duration of symptoms
- Which eye? Or both?
- Sudden or gradual?
- Metamorphopsia (wavy lines)?
- Central scotoma?
- Dark adaptation difficulty?
- Smoking history, family history, hypertension, diabetes
2. VISUAL ACUITY
- Snellen chart, each eye separately
- With best correction (glasses)
3. AMSLER GRID
- Each eye separately
- At 30 cm, with reading glasses
4. ANTERIOR SEGMENT (slit lamp)
- Rule out other causes of vision loss (cataract etc.)
5. DILATED FUNDOSCOPY
- Dilate: Tropicamide 1% + Phenylephrine 2.5%
- Look at: optic disc, macula, vessels, periphery
- Specifically: drusen (type, size, confluent?), RPE changes,
hemorrhage, exudates, elevated macula, GA, scar
6. OCT (Optical Coherence Tomography) — MANDATORY
- The most important investigation — explained in next section
Quick Check — 3 Questions
1. early, observation. 2. fundus evaluation: macular edema with subretinal hemorrhage, anti-VEGF therapy. 3. intermediate in right eye and advanced in left eye. 10-12%
- Lifestyle counselling — smoking cessation (most important), Mediterranean diet
- Amsler grid — teach home monitoring, one eye at a time daily
- Annual review — watch for progression to intermediate AMD
Early AMD → Lifestyle modification (smoking cessation, diet) + Amsler grid home monitoring + annual dilated fundoscopy. No supplements yet. No treatment needed.
- Subretinal fluid (SRF) — fluid between photoreceptors and RPE
- Intraretinal fluid (IRF) — fluid within retinal layers
- PED — RPE detachment
- Subretinal hemorrhage — if bleeding present
Urgent OCT + FFA to confirm active CNV → 3 loading doses of intravitreal anti-VEGF (ranibizumab/aflibercept/faricimab) → monthly review → Treat & Extend.
- Right eye (soft confluent drusen) = Intermediate AMD ✅
- Left eye (geographic atrophy) = Late AMD (dry) ✅
This patient has:
- Right eye: large drusen = 1 point
- Left eye: advanced AMD = 1 point
- Total person score = 2
- 5-year risk = ~12% for the right eye progressing to advanced AMD
Your Score: Good conceptual understanding, terminology needs sharpening
✅ In wet AMD — say SRF / IRF / PED, not "macular edema" ✅ 10-12%/year = fellow eye CNV risk when ONE eye already has wet AMD ✅ AREDS score = 5-year progression risk for any AMD eye
You are ready for Investigations now
- ✅ Anatomy
- ✅ Pathogenesis
- ✅ Clinical features and symptoms
- ✅ Fundoscopy signs
- ✅ Basic staging
next
PART 4 — INVESTIGATIONS: The Imaging That Drives All Decisions
THE RULE BEFORE WE START
SD-OCT is mandatory for every AMD patient at every visit. Everything else is added on top depending on what you need to know.
INVESTIGATION 1: SD-OCT (Spectral Domain Optical Coherence Tomography)
What is OCT?
How to Read an OCT — The Layers You Must Identify
VITREOUS — dark (optically empty)
━━━━━━━━━━━━━━━ Inner Limiting Membrane (bright thin line)
Nerve fiber / Ganglion layers (bright)
Inner Nuclear Layer (dark)
Outer Plexiform Layer (bright)
Outer Nuclear Layer (dark) ← thickest dark band
━━━━━━━━━━━━━━━ ELLIPSOID ZONE (EZ) — bright, very important
Inner/outer segment junction of photoreceptors
━━━━━━━━━━━━━━━ RPE — bright, continuous line at the bottom
━━━━━━━━━━━━━━━ Bruch's Membrane (just below RPE, hard to separate)
Choriocapillaris / Choroid — dark vessels
The 3 Types of Fluid on OCT — MUST KNOW
- Serous PED — pure fluid underneath, very dark space, smooth dome → often in CSCR
- Fibrovascular PED — heterogeneous (mixed bright and dark) material underneath RPE → this is wet AMD, CNV pushing RPE up
- Drusenoid PED — multiple soft drusen merging and pushing RPE up → dry AMD, high risk
OCT Signs in Dry AMD
- Drusen: Small bumps under the RPE line — the RPE line undulates up and down with each drusen
- RPE atrophy: The RPE line becomes thin, patchy, or disappears completely
- Geographic Atrophy on OCT: Complete absence of the RPE line + loss of the ellipsoid zone above it + increased signal penetrating through to the choroid (because the RPE that normally blocks light penetration is gone — this is called enhanced transmission signal or "choroidal hypertransmission")
- EZ disruption: Loss of the bright ellipsoid zone band → dead photoreceptors → irreversible vision loss
OCT Signs in Wet AMD
- SRF — dark space between photoreceptors and RPE
- IRF — dark cysts within retinal layers
- Fibrovascular PED — domed RPE with heterogeneous material below
- CNV membrane — can sometimes be seen as a hyperreflective (bright) lesion below the RPE or in the subretinal space
- Hyperreflective foci — small bright dots in the outer retina — represent migrating RPE cells or macrophages — marker of active disease
- Subretinal hyperreflective material (SHRM) — bright material in the subretinal space — blood, fibrin, or early fibrosis
INVESTIGATION 2: FFA (Fundus Fluorescein Angiography)
What is FFA?
- Choroidal flush (0-1 sec) — choroid fills first, background flush
- Arterial phase (1-3 sec) — retinal arteries fill
- Arteriovenous / venous phase (3-25 sec) — veins fill
- Late phase (5-10 min) — dye washes out from normal vessels; leaking vessels retain dye
FFA in AMD — The Two Classic CNV Patterns
- Vessels are ABOVE the RPE (in the subretinal space)
- FFA: Early, well-defined, bright hyperfluorescence in the arterial phase
- Late phase: Leakage expands beyond the original lesion margins (dye spills out)
- Looks like a lacy or wheel-spoke pattern of vessels early on
- Vessels are BELOW the RPE (sub-RPE space)
- FFA: Late, ill-defined, stippled hyperfluorescence — the RPE partially blocks the early signal
- Also called "fibrovascular PED" pattern or "late leakage of undetermined source"
- More common than classic CNV
- Classify CNV type (classic vs occult vs mixed)
- Confirm active leakage before starting anti-VEGF
- Trial eligibility (some trials required FFA-confirmed CNV)
- Detect late-stage fibrosis or atrophy
INVESTIGATION 3: ICGA (Indocyanine Green Angiography)
Why ICGA when we have FFA?
- ICGA gold standard — shows the branching inner-choroidal vascular network and the polypoidal (aneurysmal) dilations
- FFA cannot show these polyps clearly
- This distinction matters because PCV treatment (PDT + anti-VEGF) differs from typical wet AMD
- ICGA shows the "hot spot" — the plaque of vessels under the RPE that FFA cannot characterize properly
- ICGA shows the characteristic "hot spot" adjacent to a retinal vessel and the anastomosis
INVESTIGATION 4: FAF (Fundus Autofluorescence)
What is FAF?
- Geographic Atrophy = dark (hypo-AF) area — RPE is gone, no lipofuscin to fluoresce
- Stressed RPE with excess lipofuscin = bright (hyper-AF) areas — RPE is overloaded but still alive
- The edge (junctional zone) of GA — this is where FAF is most useful
FAF Junctional Zone Patterns — Predict Progression Rate
| Pattern | What it looks like | Growth rate |
|---|---|---|
| None | No increased AF at the margin | Slowest |
| Focal | 1-2 small bright spots at edge | Slow-moderate |
| Banded | Continuous bright ring around GA | Fast |
| Patchy | Scattered bright areas beyond GA | Fast |
| Diffuse | Widespread bright AF beyond GA | Fastest |
INVESTIGATION 5: OCTA (OCT Angiography)
What is OCTA?
- No dye injection (good for patients with dye allergy or renal impairment)
- Can detect non-exudative CNV — Type 1 MNV that is not leaking yet (not visible on FFA)
- Shows choriocapillaris flow deficits (predict GA progression)
- Cannot show leakage (only flow — no information about whether vessels are active/leaking)
- Artifacts from eye movement
- Cannot replace FFA for trial eligibility or leakage mapping
THE IMAGING DECISION ALGORITHM
ANY AMD PATIENT
→ SD-OCT mandatory (every visit, always)
DRY AMD / DRUSEN ONLY:
→ Add FAF (monitor GA progression, junctional zone pattern)
→ Add NIR reflectance (detect SDD — subretinal drusenoid deposits)
→ Colour fundus photo (baseline documentation)
WET AMD / FLUID ON OCT:
→ Add FFA (confirm CNV type, leakage, trial eligibility)
→ Add ICGA (if PCV suspected — Asian patient, serosanguineous PED, no drusen)
→ Add OCTA (non-invasive CNV map, choriocapillaris assessment)
Quick Check — 3 Questions
1. subretinal fluid, causing photoreceptor cells to shift causing metamorphopsia. 2. RAP or type 3 MNV, ICGA binds to proteins much longer than fluorescein which are moved faster by the choriocapillaries. 3. diffuse, frequent follow up; rapid progression
| Feature | PCV | RAP (Type 3 MNV) |
|---|---|---|
| ICGA finding | Branching network + polypoidal nodules | Hot spot adjacent to retinal vessel |
| Origin | Inner choroidal vessels | Retinal capillaries growing downward |
| Patient | Asian/African, often female | Older, bilateral, often with SDD |
| PED type | Serosanguineous, large | Small PED with retinal-choroidal anastomosis |
| Pattern | Description |
|---|---|
| Banded | Continuous ring AROUND GA margin |
| Diffuse | Widespread AF extending far BEYOND GA, not just at margin |
Your Score: 1/3 fully correct, 2/3 with fixable errors
✅ Branching network + polypoidal nodules on ICGA = PCV (not RAP) ✅ ICG penetrates RPE using near-infrared light — this is why it sees choroid ✅ Continuous ring = Banded pattern. Widespread beyond margin = Diffuse pattern
Where You Stand Now
- AREDS2 supplements — who gets them and why
- Anti-VEGF drugs — each one, how they work, the key trials
- GA treatments (the 2023 milestone drugs)
- Dosing strategies (T&E vs PRN vs fixed)
- What to say when asked about injections
next
PART 5 — TREATMENT: Everything From Supplements to Injections
THE TREATMENT MAP FIRST
EARLY AMD
→ No treatment. Lifestyle only.
INTERMEDIATE AMD
→ AREDS2 supplements + monitoring
LATE DRY AMD (Geographic Atrophy)
→ Pegcetacoplan OR Avacincaptad (FDA 2023 — first ever GA treatments)
→ Low vision rehabilitation
LATE WET AMD (Neovascular)
→ Anti-VEGF intravitreal injections — GOLD STANDARD
→ Loading × 3 → then Treat & Extend
END STAGE (bilateral, scarred)
→ Low vision aids, rehabilitation
TREATMENT 1: LIFESTYLE & PREVENTION
For ALL AMD patients and high-risk individuals:
- Oily fish (omega-3) twice a week
- Leafy green vegetables daily (kale, spinach — high in lutein and zeaxanthin)
- Nuts, olive oil
- Reduces AMD risk independently of supplements
TREATMENT 2: AREDS2 SUPPLEMENTS
The Background — Why These Supplements?
AREDS (2001) — The Original Trial
AREDS2 (2013) — The Improved Trial
- Vitamin C 500mg
- Vitamin E 400 IU
- Lutein 10mg
- Zeaxanthin 2mg
- Zinc 80mg
- Copper 2mg
WHO GETS SUPPLEMENTS? — The Critical Prescribing Rule
| AMD Stage | Supplements? |
|---|---|
| No AMD | ❌ No benefit |
| Early AMD | ❌ No benefit shown |
| Intermediate AMD | ✅ YES — start AREDS2 |
| Advanced in ONE eye | ✅ YES — protect the other eye |
| Advanced bilateral | ✅ Yes (ongoing, though limited benefit at this stage) |
TREATMENT 3: ANTI-VEGF THERAPY (Wet AMD)
The Concept — Why Anti-VEGF Works
The 6 Anti-VEGF Drugs — Know Each One
- RNA aptamer (not an antibody)
- Only blocks VEGF-A165 (one isoform)
- Given every 6 weeks
- Historically first, now obsolete — only slowed vision loss, didn't improve it
- Rarely used today
- Full IgG1 antibody, originally designed for colon cancer
- Blocks ALL VEGF-A isoforms
- 1.25 mg dose
- NOT FDA approved for eye use — used off-label
- CATT trial (2011): Non-inferior to ranibizumab
- Cost: cheapest option — very important in Nepal where cost is a major barrier
- Most widely used globally due to cost
- Fab fragment (smaller piece of antibody, 48 kDa)
- Designed specifically for the eye
- Blocks ALL VEGF-A isoforms
- 0.5 mg dose
- MARINA trial: +7.2 letters vs -10.4 sham
- ANCHOR trial: +11.3 letters vs -9.5 PDT
- These were the landmark trials that established anti-VEGF as gold standard
- Not an antibody — a fusion protein ("VEGF trap")
- Acts like a decoy receptor: has domains from both VEGFR-1 and VEGFR-2 fused to an IgG Fc
- Broader target: VEGF-A + VEGF-B + PlGF (placental growth factor)
- Higher binding affinity than ranibizumab
- 2 mg dose, given every 8 weeks after loading (fewer injections than monthly ranibizumab)
- VIEW 1&2 trial: Non-inferior to monthly ranibizumab with q8w dosing
- Single-chain antibody fragment (scFv) — smallest anti-VEGF (26 kDa)
- Highest molar concentration per injection — more drug molecules per dose
- 6 mg dose, potentially every 12 weeks
- HAWK/HARRIER: Non-inferior to aflibercept; superior fluid resolution; 56% maintained q12w
- Important caution: Post-marketing reports of intraocular inflammation and occlusive vasculitis — can cause severe vision loss. Prescribe carefully, warn patients.
- First bispecific antibody in ophthalmology
- Blocks TWO targets: VEGF-A AND Angiopoietin-2 (Ang-2)
- Why add Ang-2 blockade? Ang-2 is elevated in wet AMD and destabilizes vessels by blocking the Tie2 receptor. Blocking both VEGF-A and Ang-2 = more complete vascular stabilization
- 6 mg dose, up to every 16 weeks (longest approved interval)
- YOSEMITE/LUCERNE: 53% of patients maintained q16w dosing at 2 years
- FDA approved January 2022
Memory Aid for the Drugs in Order:
"People Buy Real Apples Before Friday" Pegaptanib → Bevacizumab → Ranibizumab → Aflibercept → Brolucizumab → Faricimab
TREATMENT 4: HOW INJECTIONS ARE GIVEN (The Procedure)
- Informed consent — explain procedure, risks, need for multiple injections
- Topical anaesthesia (proxymetacaine drops) or subconjunctival lignocaine
- Povidone iodine 5% — applied to conjunctival sac for 3 minutes (most important step to prevent endophthalmitis)
- Sterile drape, lid speculum
- Measure 3.5 mm (pseudophakic) or 4 mm (phakic) from limbus, superior or inferotemporal quadrant
- 30-gauge needle, 0.05 mL (50 µL) injection
- Check optic nerve perfusion after injection (light perception or fundoscopy)
- Post-injection: topical antibiotic drops (controversial — some centres omit)
- Warn patient: mild redness and discomfort normal. Report immediately if: severe pain, redness, vision loss → endophthalmitis
- Endophthalmitis: 0.05% per injection — most serious
- Retinal detachment: 0.01-0.04%
- RPE tear: 2-5% (with large fibrovascular PED)
- IOP spike: 30% transient — check in glaucoma patients
- Traumatic cataract: 0.01%
TREATMENT 5: DOSING STRATEGIES
Loading Phase — Always First
Then — Three Options for Maintenance:
- 3 loading → monthly review → inject only if fluid returns on OCT
- Fewer injections but undertreatment risk
- Real-world studies show PRN leads to worse outcomes than T&E
- Used mainly where resources/follow-up are difficult
- 3 loading → monthly review
- If retina is dry (no SRF/IRF): extend next interval by 2 weeks
- If retina is active (fluid present): shorten interval by 2 weeks or maintain
- Maximum extension varies by drug: q12w (ranibizumab), q16w (faricimab)
- Personalised for each patient — some patients need monthly, others can go 3-4 months
- Fewer visits than fixed monthly, better outcomes than PRN
- After loading, inject every 8 weeks regardless
- Simple, predictable
- May over-treat some and under-treat others
The Real-World Problem in Nepal:
- Patients come from distant hilly regions
- Cost of repeated injections is prohibitive
- Only a few centres offer intravitreal injections
TREATMENT 6: GA TREATMENTS (2023 — Historic)
Drug 1: Pegcetacoplan (Syfovre) — FDA February 2023
Drug 2: Avacincaptad Pegol (Izervay) — FDA August 2023
Important Honest Point:
THE TREATMENT SUMMARY TABLE
| Stage | Treatment | Key Trial/Evidence |
|---|---|---|
| Early AMD | Lifestyle only | — |
| Intermediate AMD | AREDS2 supplements | AREDS2 (2013) |
| Late Dry — GA | Pegcetacoplan or Avacincaptad | OAKS/DERBY, GATHER1 (2023) |
| Wet AMD | Anti-VEGF (ranibizumab/aflibercept/faricimab) | MARINA, ANCHOR, VIEW, YOSEMITE |
| Wet AMD extended | Brolucizumab q12w or Faricimab q16w | HAWK, YOSEMITE/LUCERNE |
| End-stage bilateral | Low vision aids, IMT, rehabilitation | — |