Adpkd
Reading File
Finding Sources
Finding Sources
Searching PubMed
"autosomal dominant polycystic kidney disease"[MeSH Terms] AND treatment
Reading File
Reading File
Reading File
Loading Image
Reading File
Excellent. I now have comprehensive content on ADPKD from multiple authoritative nephrology textbooks. Let me compile a thorough answer.
Autosomal Dominant Polycystic Kidney Disease (ADPKD)
Overview
ADPKD is the most common hereditary cystic kidney disorder, characterized by progressive, bilateral renal cyst development and enlargement, ultimately leading to chronic kidney disease (CKD) and often end-stage kidney disease (ESKD). It is a multi-system disorder with significant extrarenal manifestations.
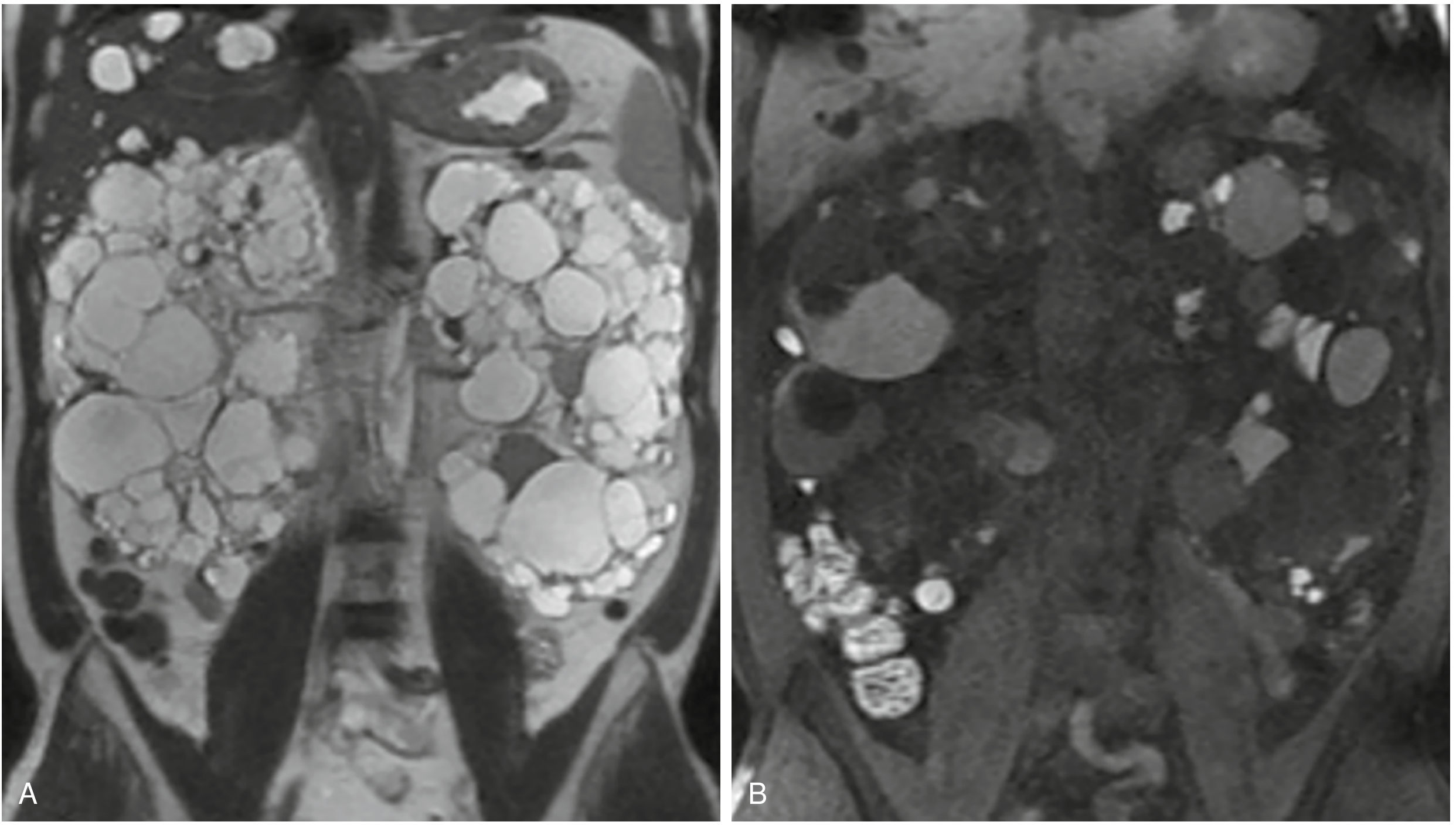
Fig. Coronal MRI of ADPKD: (A) T2-weighted — markedly enlarged kidneys with innumerable cysts, most T2 hyperintense (simple fluid), others variable. (B) T1-weighted precontrast — some cysts show T1 hyperintensity indicating proteinaceous or hemorrhagic content.
Epidemiology
- Prevalence: 1 in 400 to 1 in 1000 worldwide, in all races
- Annual ESKD incidence: ~8.7/million (men) and 6.9/million (women) in the US
- ~30,000 patients have ESKD due to ADPKD in the United States
- Men have slightly more aggressive disease (gender ratio 1.2–1.3)
- At least 10% of cases arise from apparent de novo mutations
— Brenner and Rector's The Kidney
Genetics
| Feature | Detail |
|---|---|
| Inheritance | Autosomal dominant, complete penetrance for cyst development |
| Risk per child | 50% |
| PKD1 (chr 16p13.3) | ~78% of clinical cases; mean ESKD at 58.1 years |
| PKD2 (chr 4q21) | ~15–25% of cases; mean ESKD at 79.7 years |
| Additional genes | GANAB, DNAJB11, ALG9 (atypical/milder phenotype) |
| Undetected mutations | ~7% |
- PKD1: ~65% truncating, 35% nontruncating mutations; >1,272 mutations catalogued
- PKD2: ~87% truncating, 13% nontruncating
- Homozygous PKD1/PKD2 loss-of-function → lethal in utero
- Trans-heterozygotes (one PKD1 + one PKD2 mutation) have worse disease than either alone
— Comprehensive Clinical Nephrology, 7th Ed.
Pathogenesis
Key Proteins
- Polycystin-1 (PC1) — encoded by PKD1; ~440 kDa; receptor/adhesion molecule with large extracellular N-region, 11 transmembrane domains; located in primary cilia, plasma membrane, focal adhesions, desmosomes
- Polycystin-2 (PC2) — encoded by PKD2; ~110 kDa; transient receptor potential (TRP) channel; mainly in ER, also primary cilium; forms a 1:3 complex with PC1
Two-Hit Model
Cyst formation requires somatic inactivation of the normal allele (second hit) in addition to the germline mutation — explaining why only a small fraction of tubules form cysts despite all cells carrying the germline mutation.
Downstream Signaling
Loss of polycystin function leads to:
- ↑ mTOR pathway activation (drives cell proliferation)
- ↑ cAMP (promotes fluid secretion and cell growth)
- Aberrant MAPK/ERK signaling
- Loss of primary cilium mechanosensing function
- Increased cell proliferation + apoptosis
Pathology
- Cysts arise from all nephron segments, predominantly distal tubule and collecting duct
- Cysts grow, dissociate from parent tubule, becoming isolated fluid-filled sacs
- Polycystic kidneys show: afferent arteriole/interlobular artery sclerosis, interstitial fibrosis, tubular hyperplasia — even when GFR is normal
- Histology: papillary hyperplasia of cyst epithelium; microscopic adenomas present (risk of RCC is not increased)
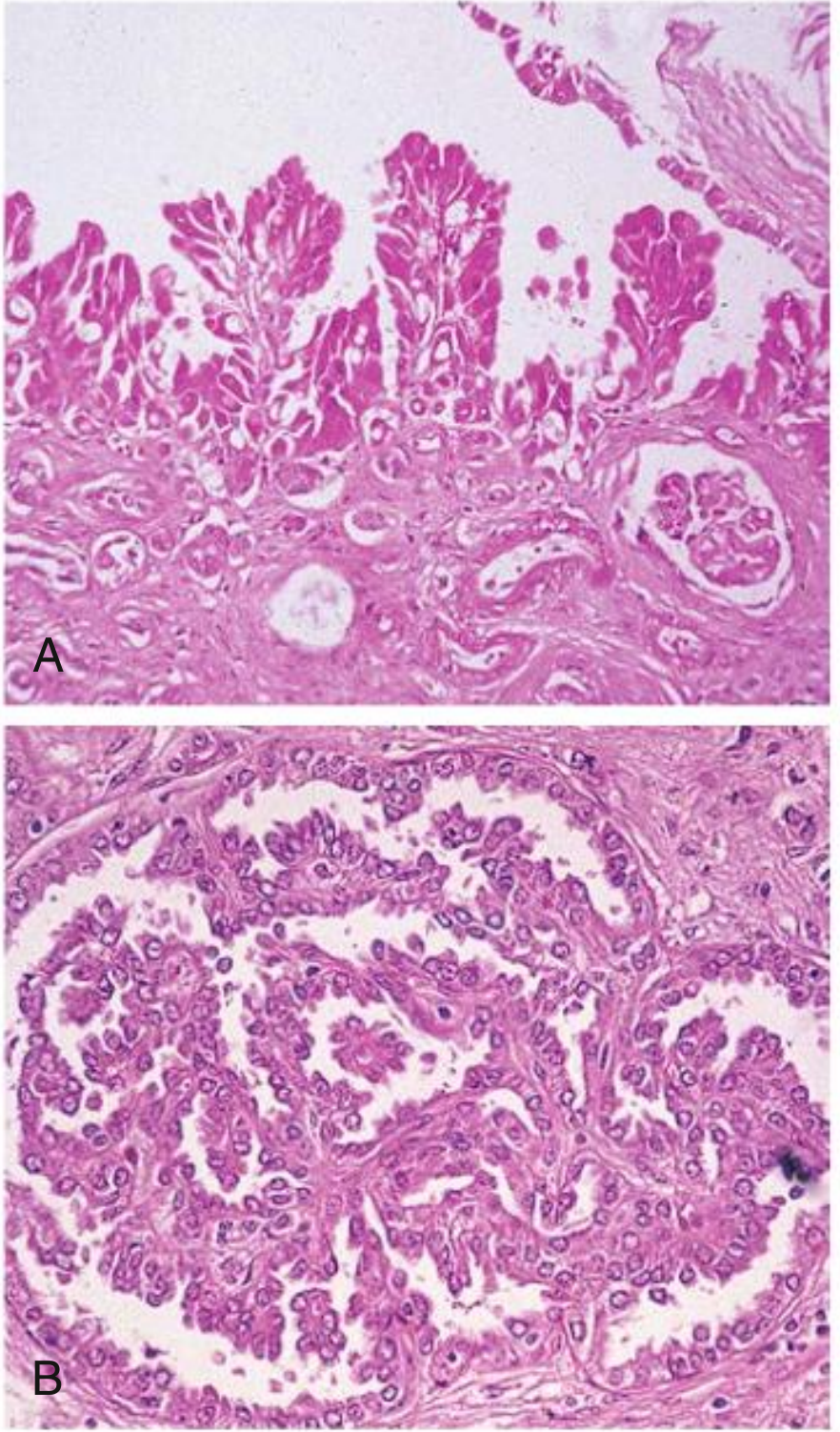
Clinical Features
Renal
- Flank pain / abdominal pain — most common symptom; due to cyst enlargement, hemorrhage, infection, or stone
- Hematuria — gross or microscopic; intracyst hemorrhage is most common cause
- Hypertension — affects >50% early in disease, often precedes GFR decline; driven by intrarenal RAAS activation
- Nephrolithiasis — ~20% of patients; predominantly uric acid and calcium oxalate stones (associated with hypocitraturia, low urine pH)
- Cyst infection — presents with fever, flank/loin pain, elevated CRP; difficult to distinguish from hemorrhage; trimethoprim-sulfamethoxazole or fluoroquinolones (lipophilic agents penetrate cysts)
- CKD progression → ESKD — median age of ESKD: 58.1 (PKD1), 79.7 (PKD2)
Extrarenal
| Organ | Manifestation |
|---|---|
| Liver | Polycystic liver disease (most common extrarenal finding); more severe in women of childbearing age |
| Cardiovascular | Mitral valve prolapse (~25%); intracranial aneurysms (~8–12%; ruptured SAH risk) |
| Intracranial | Berry aneurysms — screen if family history of aneurysm/SAH or high-risk occupation |
| Pancreas | Pancreatic cysts (rare, usually asymptomatic) |
| Seminal vesicles | Cysts |
| Arachnoid membranes | Cysts |
Diagnosis
Ultrasound Criteria (Pei/Ravine, unified criteria)
| Age | With family history | Without family history |
|---|---|---|
| < 40 years | ≥ 3 cysts bilaterally | — |
| 40–60 years | ≥ 4 cysts bilaterally | — |
| > 60 years | ≥ 8 cysts bilaterally | — |
| Any age | — | ≥ 20 bilateral cysts |
Imaging
- Ultrasound: first-line; limited by body habitus and large kidney size
- MRI: preferred for monitoring disease progression and identifying solid masses among cysts; superior soft tissue contrast
- CT: good for complications (hemorrhage, infection, malignancy)
- Total Kidney Volume (TKV): key prognostic biomarker; measured by MRI or CT; Mayo Clinic Imaging Classification (MCIC) uses ellipsoid formula for practical risk stratification
Genetic Testing
- Indicated when imaging is equivocal, for living-related donor evaluation, or pre-implantation genetic diagnosis
- Next-generation sequencing (NGS) of PKD1/PKD2 is current standard; PKD1 screening is complex due to segmental duplication
Prognosis / Risk Stratification
Mayo Clinic Imaging Classification (MCIC) classifies patients by height-adjusted TKV (htTKV) into 5 classes (1A–1E), with 1C–1E having rapidly progressive disease (>5 ml/cm/year kidney growth).
PROPKD score (genetic + clinical factors) also helps predict time to ESKD.
Treatment
General / Supportive
- Adequate hydration — high water intake (2.5–3 L/day) suppresses vasopressin/AVP, reducing cAMP-driven cyst growth; goal urine osmolality <280 mOsm/kg
- Hypertension control: RAAS blockade with ACE inhibitors or ARBs first-line; target BP <110/75 mmHg in young patients (HALT-PKD trial data)
- Low-sodium, low-protein diet (standard CKD measures)
- Avoid nephrotoxins: NSAIDs, contrast agents
- Statin therapy: may have modest benefit; used for cardiovascular risk reduction
Pain Management
- Exclude reversible causes (infection, stone, tumor)
- Conservative first: reassurance, lifestyle modification, tricyclic antidepressants
- Narcotic analgesics for acute severe pain only (avoid long-term)
- Cyst decompression: aspiration ± sclerotherapy (95% ethanol, minocycline, sodium tetradecyl sulfate) for large symptomatic cysts; laparoscopic or surgical cyst unroofing for refractory cases
Disease-Modifying / Novel Therapies
| Drug | Mechanism | Status |
|---|---|---|
| Tolvaptan (Jynarque) | V2 receptor antagonist → ↓ cAMP → slows cyst growth | FDA-approved (2018); indicated for adults with rapidly progressing ADPKD (MCIC 1C–1E or equivalent) |
| mTOR inhibitors (sirolimus, everolimus) | ↓ mTOR signaling | Shown to reduce kidney growth in trials but did not preserve GFR; not routinely recommended |
| Somatostatin analogues (octreotide, lanreotide) | ↓ cAMP | Slow liver and kidney cyst growth; trials ongoing |
Tolvaptan key points:
- TEMPO 3:4 and REPRISE RCTs showed reduced TKV growth and slowing of GFR decline
- Main side effects: aquaretic (polyuria, polydipsia, nocturia), hepatotoxicity (requires liver function monitoring; REMS program in US)
- Contraindicated in patients who cannot sense/respond to thirst
Cyst Infection
- Fluoroquinolones or TMP-SMX (lipophilic → penetrate cysts)
- Prolonged treatment (4–6 weeks) often required
ESKD Management
- Dialysis or kidney transplantation
- Transplantation outcomes are excellent; pre-emptive transplant preferred
- Native nephrectomy may be needed for massively enlarged kidneys prior to transplant (space issue)
Intracranial Aneurysm Screening
- Prevalence ~8–12% in ADPKD (vs. 2–3% general population)
- Screen with MRA (without gadolinium) in:
- Family history of intracranial aneurysm or SAH
- Prior aneurysm rupture
- High-risk occupation (pilots, surgeons) or preparing for elective surgery under GA
- Significant anxiety about risk
- If no aneurysm found and no family history: routine screening not generally recommended
Summary
ADPKD is a progressive, systemic ciliopathy caused by mutations in PKD1 or PKD2, leading to bilateral renal cyst accumulation, ESKD by the sixth decade (PKD1) or later (PKD2), and important extrarenal complications including polycystic liver disease and intracranial aneurysms. Management centers on vasopressin suppression, aggressive BP control, and — for rapidly progressive disease — tolvaptan, the only approved disease-modifying therapy.
Sources: Comprehensive Clinical Nephrology, 7th Ed. | NKF Primer on Kidney Diseases, 8th Ed. | Brenner and Rector's The Kidney | Grainger & Allison's Diagnostic Radiology
Medullary kidney disease
Finding Sources
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Finding Sources
Reading File
Searching Images
medullary sponge kidney nephronophthisis medullary cystic kidney disease histology imaging
I now have comprehensive content from Harrison's, Goldman-Cecil, NKF Primer, Robbins, and Comprehensive Clinical Nephrology. Let me compile the full answer.
Medullary Kidney Disease
"Medullary kidney disease" encompasses a spectrum of distinct conditions whose primary pathology involves the renal medulla. The major entities are:
- Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD) — formerly "Medullary Cystic Kidney Disease" (MCKD)
- Nephronophthisis (NPHP) — the childhood ciliopathy
- Medullary Sponge Kidney (MSK) — a structural collecting duct anomaly
1. Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD)
Nomenclature
The term ADTKD (KDIGO consensus, 2015) has officially replaced "medullary cystic kidney disease" because:
- Cysts are not invariably present (especially early disease)
- Histology shows predominantly tubulointerstitial fibrosis, not cystic change
- The old MCKD grouping incorrectly lumped it with nephronophthisis, which is a distinct entity
Genetic Subtypes
| Subtype | Gene | Chr | Key Features |
|---|---|---|---|
| ADTKD-UMOD (MCKD2 / FJHN) | UMOD | 16p12 | Most common (~50%); uromodulin (Tamm-Horsfall protein) mutation; hyperuricemia + gout; also called uromodulin-associated kidney disease (UAKD) |
| ADTKD-MUC1 (MCKD1) | MUC1 | 1q21 | ~30% of cases; toxic neoprotein fragment; no hyperuricemia early; late gout only from CKD |
| ADTKD-REN | REN | 1q32.1 | Rare; hyporeninemia → hyperkalemia + anemia (↓ Ang II → ↓ erythropoiesis); early hyperuricemia |
| ADTKD-HNF1B | HNF1B | 17q12 | Multi-organ: kidney, liver, pancreas, genital tract; variable severity |
| ADTKD-SEC61A1 | SEC61A1 | — | Rarest; ER translocon mutation |
Pathogenesis — ADTKD-UMOD (Most Common)
Uromodulin is expressed on the luminal side of thick ascending limb (TAL) and early DCT. Mutations impair trafficking of the furosemide-sensitive Na-K-2Cl transporter to the apical membrane → mild sodium wasting → volume contraction → ↑ proximal urate reabsorption → hyperuricemia. Intracellular accumulation of mutant uromodulin triggers tubular cell death. UMOD mutations are found in 2–3% of adults with CKD.
Clinical Features
- Onset: typically 4th–7th decade (except FJHN and GCKD, allelic variants presenting in the 1st–3rd decade)
- Slowly progressive CKD with bland urinary sediment — minimal proteinuria, no hematuria
- Hyperuricemia + gout (ADTKD-UMOD, ADTKD-REN; less prominent in ADTKD-MUC1 early on)
- Anemia disproportionate to GFR in ADTKD-REN (↓ Ang II → ↓ erythropoiesis)
- No increased UTI or nephrolithiasis
Investigations
- Urinalysis: bland sediment, minimal or no proteinuria
- Labs: hyperuricemia, ↓ uric acid fractional excretion, mild ↓ urinary concentrating ability
- Imaging: normal to mildly reduced kidney size, ↑ echogenicity, loss of corticomedullary differentiation; medullary cysts visible on US/CT — but may be too small to detect, especially early
- Biopsy: tubulointerstitial fibrosis, tubular atrophy, tubular BM disintegration — not diagnostic of specific subtype
- Genetic testing required for definitive diagnosis (NGS panels available)
Treatment
- Allopurinol or febuxostat for hyperuricemia/gout
- Management of CKD sequelae: anemia, acidosis, MBD, hypertension
- HNF1B: manage pancreatic insufficiency
- ADTKD-REN: manage hypotension and hyperkalemia
- No disease-specific therapy exists; no recurrence after kidney transplantation
- ESKD: age 40–70 years for ADTKD (cf. NPHP, below)
— Harrison's 22E | Goldman-Cecil Medicine | NKF Primer on Kidney Diseases, 8e | Comprehensive Clinical Nephrology, 7e
2. Nephronophthisis (NPHP)
Overview
NPHP is a large family of autosomal recessive ciliopathies — the most common genetic cause of ESKD in children and young adults. >90 genes identified. Must be distinguished from ADTKD despite historical confusion.
Genetics
| Feature | Details |
|---|---|
| Inheritance | Autosomal recessive |
| Loci | NPHP1–NPHP20+ (nephrocystins); mutations in >90 genes |
| Most common (juvenile) | NPHP1 deletion (~20% of cases) |
| Infantile form | NPHP2 (inversin) mutations |
Nephrocystin proteins localize to: primary cilia, basal bodies, centrosomes, mitotic spindles — all components of the cilium/centrosome axis. Defects disrupt planar cell polarity (PCP) and tubular epithelial maintenance.
Genetic Table (Key Forms)
| Form | Gene | Protein | Extrarenal Manifestations |
|---|---|---|---|
| NPHP1 | NPHP1 | Nephrocystin-1 | Retinitis pigmentosa, oculomotor apraxia |
| NPHP2 (infantile) | INVS | Inversin | Situs inversus, liver fibrosis, retinitis pigmentosa |
| NPHP3 | NPHP3 | Nephrocystin-3 | Liver fibrosis, Meckel-Gruber syndrome |
| NPHP5 | IQCB1 | Nephrocystin-5 | Retinitis pigmentosa (all cases) |
| NPHP6 | CEP290 | Nephrocystin-6 | Retinitis pigmentosa, Joubert syndrome, Meckel-Gruber |
| NPHP11 | TMEM67 | Meckelin | Retinitis pigmentosa, polydactyly, liver fibrosis |
— NKF Primer on Kidney Diseases, 8e
Clinical Variants
- Infantile NPHP (NPHP2): ESKD in early childhood
- Juvenile NPHP (NPHP1, most common): ESKD by ~13 years; polyuria and polydipsia as first symptoms
- Adolescent NPHP: ESKD in early adulthood
Associated Syndromes
| Syndrome | Features |
|---|---|
| Senior-Loken syndrome | NPHP + retinitis pigmentosa |
| Joubert syndrome | NPHP + cerebellar vermis hypoplasia ("molar tooth" on MRI) + hyperpnea/apnea |
| Bardet-Biedl syndrome (BBS) | NPHP-like kidney + truncal obesity, cognitive impairment, retinal dystrophy, polydactyly, hypogonadism; ≥26 BBS genes |
Clinical Features
- Polyuria + polydipsia — earliest symptoms; severe urinary concentrating defect
- Sodium wasting + tubular acidosis
- Bland urinalysis — no proteinuria, no hematuria
- Hypertension — late finding (contrast with ADPKD)
- Progressive CKD → ESKD within 5–10 years of onset
Pathology
- Kidneys: small, contracted granular surface
- Medullary/corticomedullary cysts (1–15 mm); cysts also in cortex
- Histology: tubular BM thickening and disruption, tubular atrophy, interstitial fibrosis, inflammatory infiltrate; glomeruli relatively preserved early
Key pathological distinction: medullary cysts are present, but it is the cortical tubulointerstitial damage that drives renal failure.
Diagnosis
- Medullary cysts may be too small to visualize radiographically
- Suspect in: child/adolescent with unexplained CKD + positive family history + tubulointerstitial nephritis on biopsy
- Genetic panel sequencing is definitive (NGS available clinically)
Treatment
- No curative or targeted therapy exists; no clinical trials for NPHP-specific interventions
- Manage CKD complications: anemia, acidosis, electrolyte imbalance, MBD, growth retardation
- Kidney transplantation — successful, no recurrence; eventually required for most patients
3. Medullary Sponge Kidney (MSK)
Definition
MSK is a congenital, non-progressive structural anomaly characterized by multiple cystic dilations of inner medullary and papillary collecting ducts, giving a spongy gross appearance to the medulla.
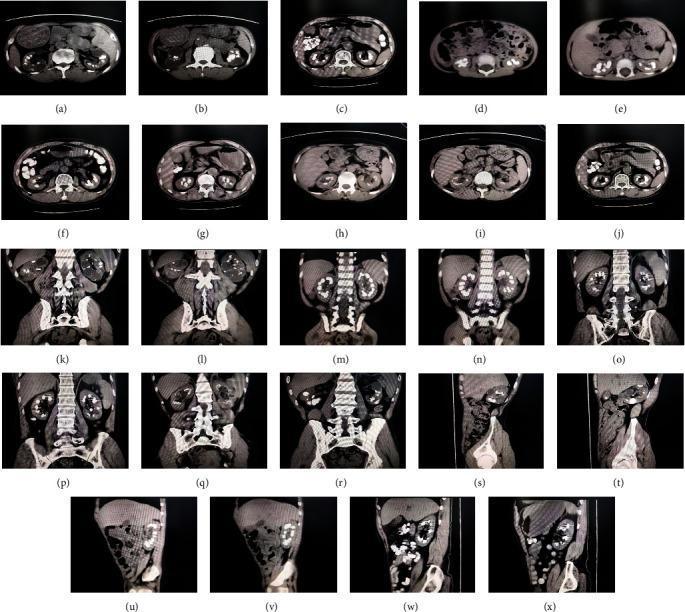
CT showing bilateral medullary nephrocalcinosis and papillary collecting duct ectasia characteristic of MSK
Epidemiology
- General population frequency: ~1 in 5000 (likely underestimated; many asymptomatic)
- Up to 20% of patients with nephrolithiasis have at least mild MSK
- Usually sporadic; rare autosomal dominant familial cases reported
- Associated with: congenital hemihypertrophy, Beckwith-Wiedemann syndrome, CAKUT, Wilms tumor
Pathogenesis
A developmental defect in ureteric bud–metanephric mesenchyme interaction (evidence: RET proto-oncogene and GDNF gene defects; embryonal tissue in affected papillae).
Clinical Features
- Often asymptomatic — incidental finding
- Symptoms in 2nd–3rd decade (sometimes 4th–5th decade)
- Recurrent nephrolithiasis — calcium phosphate (apatite) ± calcium oxalate stones
- Hematuria (microscopic or gross; may be unrelated to stones)
- Recurrent UTI
- Urinary concentrating defect + impaired urinary acidification (incomplete distal RTA)
- Bone mineralization defects from chronic acidification abnormalities
- Renal function usually normal; ESKD is uncommon
Pathology
- Pathology confined to medullary and intrapapillary collecting ducts
- Cysts 1–8 mm (spherical/oval); may contain apatite concretions
- Affected pyramids and calyces enlarged; cortex normal
- Cysts lined by cuboidal or transitional epithelium
Diagnosis
- Plain X-ray: radiopaque medullary concretions (nephrocalcinosis)
- CT urography or IVU: characteristic "bouquet of flowers" / "paintbrush" pooling of contrast in ectatic medullary ducts — historically the gold standard
- Differential: hyperparathyroidism, distal RTA type I, hypervitaminosis D, milk-alkali syndrome, sarcoidosis (all can cause medullary nephrocalcinosis)
Treatment & Prognosis
- Control stone formation: high fluid intake, thiazide diuretics (for hypercalciuria), potassium citrate (for hypocitraturia/acidification defect)
- Treat UTIs promptly
- Long-term prognosis excellent; ESKD is rare
Comparison Table
| Feature | ADTKD (MCKD) | Nephronophthisis | Medullary Sponge Kidney |
|---|---|---|---|
| Inheritance | Autosomal dominant | Autosomal recessive | Usually sporadic |
| Age of onset | 4th–7th decade | Childhood/adolescence | 2nd–4th decade |
| ESKD | 40–70 years | 5–15 years (juvenile) | Rare |
| Key labs | Hyperuricemia, bland UA | Bland UA, hyposthenuria | Normal or ↓ acidification |
| Hypertension | Late/mild | Late finding | Absent |
| Proteinuria | Minimal | Absent/minimal | Absent |
| Cyst location | Corticomedullary | Corticomedullary | Papillary collecting ducts only |
| Extrarenal | Gout (UMOD); pancreatic/genital (HNF1B) | Retinal, cerebellar, liver | Hemihypertrophy, BW syndrome |
| Pathogenesis | Ciliary/ER protein misfolding | Ciliopathy | Developmental ureteric bud defect |
| Treatment | Allopurinol; CKD Mx | CKD Mx; transplant | Stone prevention; excellent prognosis |
Sources: Harrison's Principles of Internal Medicine 22E | Goldman-Cecil Medicine | NKF Primer on Kidney Diseases, 8e | Comprehensive Clinical Nephrology, 7e | Robbins & Cotran Pathologic Basis of Disease
Medullary kidney disease
The previous response already covered Medullary Kidney Disease comprehensively. Here's a concise summary of what was covered — let me know if you want to go deeper on any specific aspect:
Medullary Kidney Disease — Summary
Three distinct entities:
1. ADTKD (formerly Medullary Cystic Kidney Disease)
- AD inheritance, adult onset (4th–7th decade)
- Genes: UMOD (most common, hyperuricemia/gout), MUC1, REN, HNF1B, SEC61A1
- Bland urinalysis, slowly progressive CKD → ESKD age 40–70
- Diagnosis by genetic testing; no disease-specific therapy
2. Nephronophthisis (NPHP)
- AR ciliopathy, >90 genes, childhood/adolescent onset
- Most common genetic cause of ESKD in children
- Polyuria/polydipsia earliest symptoms; bland urine; hypertension is late
- Syndromes: Senior-Loken (+ retinitis pigmentosa), Joubert (+ cerebellar vermis hypoplasia), Bardet-Biedl
- No curative therapy; kidney transplant eventually required
3. Medullary Sponge Kidney (MSK)
- Sporadic, collecting duct ectasia, papillary calcifications
- Recurrent stones (calcium), hematuria, UTI; concentrating/acidification defect
- Renal function usually preserved; ESKD rare
- Treatment: hydration, citrate, thiazides
Would you like to focus on any one of these — e.g., genetics, clinical management, differentiating features, or associated syndromes?
Cystic kidney disease
Your credits are exhausted. Your daily credits reset tomorrow, or you can purchase a top-up for more.
This is a shared conversation. Sign in to Orris to start your own chat.