Pathophysiology of rickets
Reading File
Finding Sources
Searching PubMed
rickets pathophysiology vitamin D deficiency
Reading File
Reading File
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
I now have comprehensive information from multiple authoritative textbooks. Let me compile the full answer.
Pathophysiology of Rickets
Rickets is a disorder of impaired bone mineralization at the growth plate in growing children, resulting from an insufficient calcium-phosphate product in the extracellular fluid. The same process in adults (after growth plate closure) is called osteomalacia.
The Vitamin D/Calcium/Phosphate Pathway
Normal bone mineralization requires:
- An adequate supply of Ca²⁺ and PO₄³⁻ from the extracellular fluid
- An appropriate pH (~7.6)
- Normal bone matrix composition
- Control of inhibitors of mineralization
Vitamin D is activated by a two-step hydroxylation:
- Step 1 - Liver: D₃ → 25-hydroxyvitamin D₃ [25(OH)D₃]
- Step 2 - Kidney: 25(OH)D₃ → 1,25-dihydroxyvitamin D₃ [1,25(OH)₂D₃ = calcitriol], catalyzed by renal 1α-hydroxylase
Without activated vitamin D, only 10-15% of dietary calcium and ~60% of phosphorus are absorbed from the gut (compared to much higher absorption with normal vitamin D). - Bailey & Love's Short Practice of Surgery, p. 1909
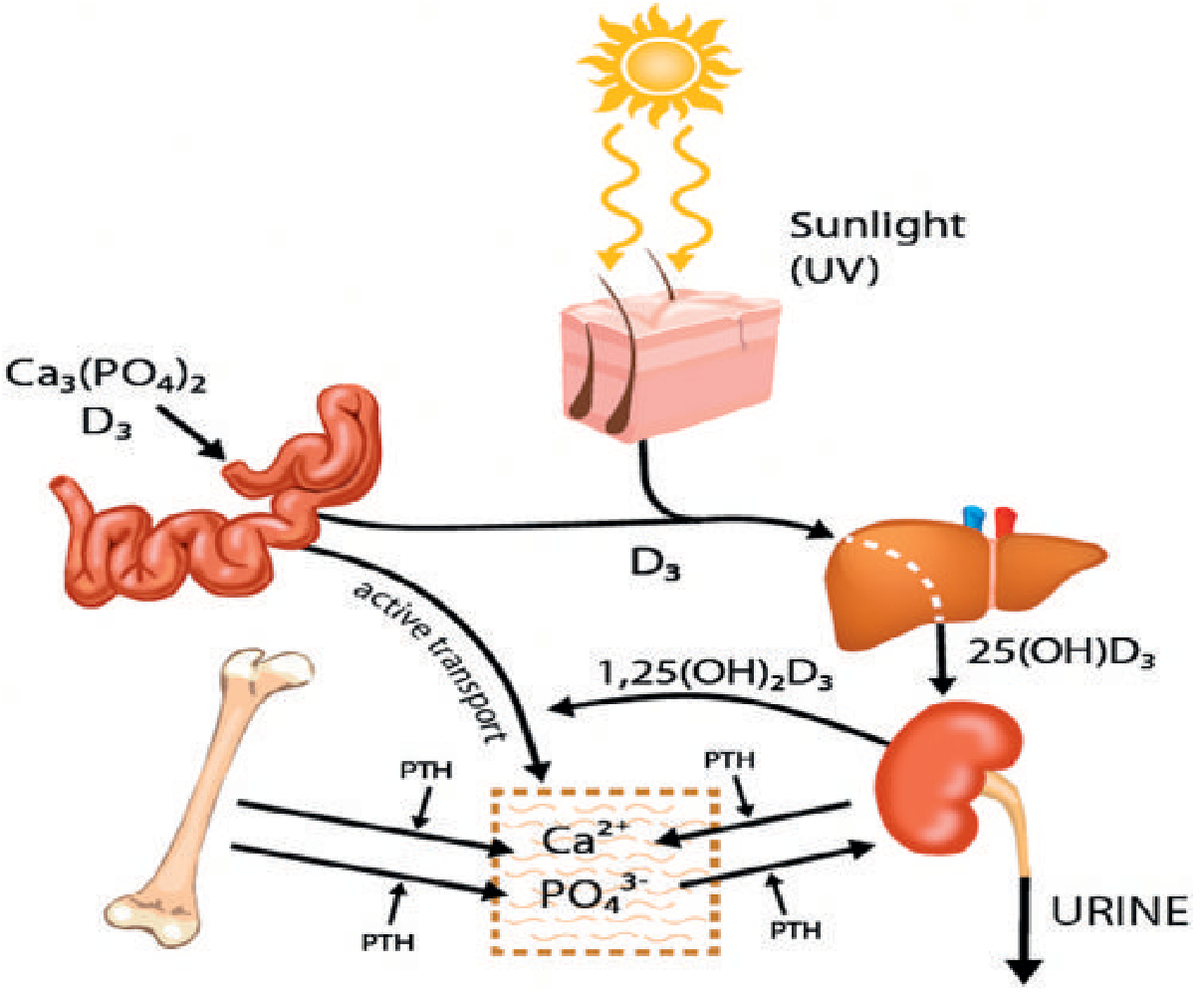
Core Pathophysiological Cascade
1. Reduced Intestinal Absorption of Ca²⁺ and PO₄³⁻
When vitamin D is deficient, intestinal absorption falls sharply. This causes a transient fall in serum ionized calcium, sensed by the parathyroid glands. - Rheumatology, 2-Volume Set, p. 1859
2. Secondary Hyperparathyroidism (SHPT)
The parathyroid glands respond to low serum Ca²⁺ by increasing PTH secretion. PTH:
- Stimulates osteoclastic bone resorption - releasing Ca²⁺ from bone to maintain near-normal serum calcium
- Increases renal Ca²⁺ reabsorption
- Reduces renal phosphate reabsorption - causing phosphaturia and hypophosphatemia
This is why in rickets, serum calcium is only slightly depressed (PTH compensates), but serum phosphate is greatly depressed (PTH actively promotes urinary phosphate loss). - Guyton & Hall Medical Physiology, p. 994
3. Failure to Mineralize Osteoid
- The resulting low calcium-phosphate product is insufficient to mineralize osteoid laid down by osteoblasts
- Large amounts of unmineralized osteoid accumulate - widened osteoid seams on histology
- Important note: vitamin D itself is not strictly required for osteoid mineralization. The key problem is failure to deliver adequate Ca²⁺ and PO₄³⁻ to mineralization sites. Evidence: patients with VDR mutations maintained on high Ca²⁺/PO₄³⁻ infusions do mineralize their osteoid normally. - Rheumatology, p. 1858
4. Growth Plate Disruption (Specific to Rickets)
In a normal growth plate:
- Chondrocytes in the resting zone mature into columns of proliferating chondrocytes
- These hypertrophy and undergo apoptosis (essential for the orderly scaffold)
- Calcified cartilage is replaced by mineralized bone (endochondral ossification)
In rickets, low extracellular Ca²⁺ and PO₄³⁻ impair hypertrophic chondrocyte apoptosis. The chondrocyte scaffold is not cleared, leading to:
- An expanded, disorganized, hypomineralized growth plate
- An enlarged and distorted zone of hypertrophy (maturation zone)
- A poorly defined zone of provisional calcification
- Growth arrest and short stature
This produces the classic radiological appearances: widened, flared, and cupped metaphyses with indistinct epiphyseal margins. - Grainger & Allison's Diagnostic Radiology, p. 1909; Miller's Review of Orthopaedics, p. 322
5. Progressive Bone Weakening
With prolonged PTH elevation:
- Osteoclastic resorption intensifies
- Osteoblasts respond by laying down more osteoid - but this osteoid fails to calcify
- Uncalcified, weak osteoid progressively replaces resorbed bone
- Result: bowing of weight-bearing bones (genu varum typically), pathological fractures (Looser zones/pseudofractures), and bone pain - Guyton & Hall, p. 994
6. Tetany (in severe or late cases)
Once calcium stores are depleted from bone, serum Ca²⁺ falls rapidly. When it drops below 7 mg/dL, tetany develops (carpopedal spasm, laryngospasm). Tetany is uncommon early because PTH compensates, but is a late and potentially lethal complication. - Guyton & Hall, p. 994
Biochemical Summary
| Parameter | Change in Rickets | Reason |
|---|---|---|
| Serum Ca²⁺ | Low-normal or slightly low | PTH compensates by releasing bone Ca²⁺ |
| Serum PO₄³⁻ | Markedly low | PTH drives urinary phosphate loss |
| PTH | Elevated | Secondary hyperparathyroidism |
| Alkaline Phosphatase (ALP) | Elevated | Increased osteoblastic activity producing unmineralized osteoid |
| 25(OH)D₃ | Low (in nutritional rickets) | Deficient substrate |
| 1,25(OH)₂D₃ | Low | Deficient or not produced |
- Tietz Textbook of Laboratory Medicine, p. 2210; Henry's Clinical Diagnosis, p. 259
Histological Findings
- Widened osteoid seams (unmineralized bone matrix)
- "Swiss cheese" trabeculae
- Enlarged, distorted hypertrophic zone of growth plate
- Irregular tetracycline uptake on bone biopsy (patchy, blurred) - Miller's Review of Orthopaedics, p. 322; Henry's Clinical Diagnosis, p. 259
Types of Rickets and Their Specific Pathophysiology
| Type | Mechanism |
|---|---|
| Nutritional (Vitamin D deficiency) | Inadequate sunlight or dietary vitamin D → deficient substrate for hydroxylation |
| Malabsorptive | Fat-soluble vitamin D not absorbed (celiac, biliary/pancreatic disease, steatorrhea) |
| Renal (renal rickets/renal osteodystrophy) | Damaged kidneys fail to convert 25(OH)D₃ to 1,25(OH)₂D₃ via 1α-hydroxylase |
| Hereditary Vit D-Dependent Rickets Type I (VDDR-I) | Inactivating mutation in renal 1α-hydroxylase (CYP27B1) gene (AR, chr 12q14) - no active vitamin D produced; responds to calcitriol but not plain vitamin D |
| Hereditary Vit D-Dependent Rickets Type II (VDDR-II) | Inactivating mutation in the VDR gene - end-organ resistance; no response to either vitamin D or calcitriol |
| X-linked Hypophosphatemic Rickets (XLH) | X-linked dominant; mutation in PHEX (phosphate-regulating endopeptidase) → elevated FGF-23 → reduced renal phosphate reabsorption and impaired renal 1α-hydroxylase → isolated hypophosphatemia with normal Ca²⁺; first-line treatment: burosumab (anti-FGF-23 monoclonal antibody) |
| Tumor-Induced Osteomalacia (Oncogenic Rickets) | Mesenchymal tumor overproduces FGF-23 → phosphaturia, hypophosphatemia |
| Calcium-deficiency rickets | Primarily low dietary calcium (even with adequate vitamin D) - common in certain African populations |
| Fanconi syndrome / Renal tubular acidosis | Generalized proximal tubular dysfunction → phosphaturia, systemic acidosis → impaired mineralization |
- Miller's Review of Orthopaedics; Henry's Clinical Diagnosis; Rheumatology 2022; Grainger & Allison's Radiology
Key Clinical Manifestations (Mechanistic Link)
- Bowed legs (genu varum): Softened, unmineralized bone bends under weight-bearing
- Rachitic rosary: Expanded, unmineralized costochondral junctions
- Craniotabes / frontal bossing: Soft skull bones; delayed fontanelle closure
- Harrison sulcus: Chest wall deformity from respiratory muscle pull on softened ribs
- Dental defects: Disrupted enamel and dentin mineralization
- Muscle hypotonia: Linked to hypophosphatemia and low 1,25(OH)₂D₃ effects on muscle
- Waddling gait / short stature: Growth plate failure and bowing deformities
Recent literature note: A 2026 review (PMID 41741919) covers refractory rickets evaluation and management, and a 2025 review (PMID 40295317) covers XLH diagnosis and therapy - consistent with the mechanisms described above, with no new contradicting evidence.
What is the thing that occurs at the growth plate
Based on what was already retrieved in our previous search, I can answer this directly from the textbook content.
What Happens at the Growth Plate in Rickets
Normal Growth Plate (Endochondral Ossification)
The growth plate has organized zones:
- Resting zone - stem chondrocytes in reserve
- Proliferative zone - chondrocytes divide and arrange into longitudinal columns
- Hypertrophic zone (maturation zone) - chondrocytes enlarge
- Zone of provisional calcification - hypertrophied chondrocytes undergo apoptosis, the cartilage matrix calcifies, and osteoclasts/osteoblasts replace it with mineralized bone
The key step is chondrocyte apoptosis - it clears the scaffold so mineralized bone can be laid down in an orderly fashion.
What Goes Wrong in Rickets
Low extracellular Ca²⁺ and PO₄³⁻ impair hypertrophic chondrocyte apoptosis.
The sequence:
- Hypertrophic chondrocytes fail to die on schedule
- The cartilage scaffold is not cleared
- Columns of chondrocytes accumulate - the hypertrophic zone expands dramatically
- The zone of provisional calcification is poorly defined or absent
- The result is a disorganized, expanded, hypomineralized growth plate
This directly causes the classic radiological findings:
- Widened, flared, cupped metaphyses
- Indistinct epiphyseal margins
- Physeal widening on X-ray
And clinically: growth retardation and short stature, because the machinery of longitudinal bone growth is broken.
Why phosphate specifically matters here
Phosphate (inorganic phosphate, Pi) is a direct signal for chondrocyte apoptosis. When serum phosphate is low (from PTH-driven phosphaturia), chondrocytes in the hypertrophic zone do not receive the signal to undergo programmed cell death. This is why hypophosphatemia is the dominant driver of the growth plate lesion - even more so than hypocalcemia. - Rheumatology, 2-Volume Set (Elsevier 2022), p. 1859
In one sentence: In rickets, low phosphate blocks hypertrophic chondrocyte apoptosis, causing the growth plate's hypertrophic zone to expand into a wide, disorganized, unmineralized mass - which is what you see as the characteristic "cupped and frayed" metaphysis on X-ray.
This is a shared conversation. Sign in to Orris to start your own chat.