Explain the mechanism of acute respiratory distress syndrome
Reading File
Finding Sources
Searching PubMed
"acute respiratory distress syndrome"[MeSH Terms] AND pathogenesis
Reading File
Reading File
Finding Sources
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
Loading Image
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Now I have comprehensive information from authoritative textbooks. Let me compile a detailed, well-structured answer.
Acute Respiratory Distress Syndrome (ARDS): Mechanism
Overview
ARDS is a syndrome of acute, diffuse, inflammatory lung injury that leads to increased pulmonary vascular permeability, loss of aerated lung tissue, and refractory hypoxemia. It develops in response to either direct (pulmonary) or indirect (extrapulmonary) insults, and its pathologic hallmark is diffuse alveolar damage (DAD). Mortality routinely exceeds 30%.
Common triggers:
- Direct: Pneumonia, aspiration of gastric contents, pulmonary contusion, inhalation injury
- Indirect: Sepsis (most common), pancreatitis, major trauma, transfusion-related lung injury (TRALI), burns
Phases of ARDS

1. Exudative Phase (Days 1-7)
The initiating event is injury to the alveolar-capillary barrier, which has two critical components:
A) Alveolar epithelial injury
- Type I pneumocytes (which cover ~95% of the alveolar surface) are particularly vulnerable. Their death disrupts barrier integrity, allows flooding of the alveolar space with proteinaceous fluid, and eliminates the cells responsible for alveolar fluid clearance.
- Type II pneumocytes are also damaged, impairing surfactant synthesis.
- Multiple mechanisms drive epithelial cell death: necrosis, apoptosis, coagulation cascade activation, and mechanical stretch.
- Loss of functional ENaC (epithelial Na+ channels) and Na+/K+-ATPase on alveolar epithelial cells impairs active sodium - and thus fluid - reabsorption from the alveolar space, perpetuating edema.
B) Microvascular endothelial injury
- Loss of pulmonary vascular endothelial barrier integrity is both necessary and sufficient for ARDS development.
- Disruption of tight junctions and adherens junctions allows plasma proteins and fluid to leak into the interstitium and alveoli.
- Elevated angiopoietin-2 (Ang2) - released from endothelial cells - competitively antagonizes the barrier-stabilizing angiopoietin-1/Tie2 axis, amplifying vascular leak. Genetic variants in Ang2 are associated with higher ARDS risk.
The net result is flooding of alveoli with protein-rich, inflammatory fluid, forming the characteristic hyaline membranes (fibrin + cell debris) seen histologically.
The Role of Neutrophils and Inflammatory Mediators
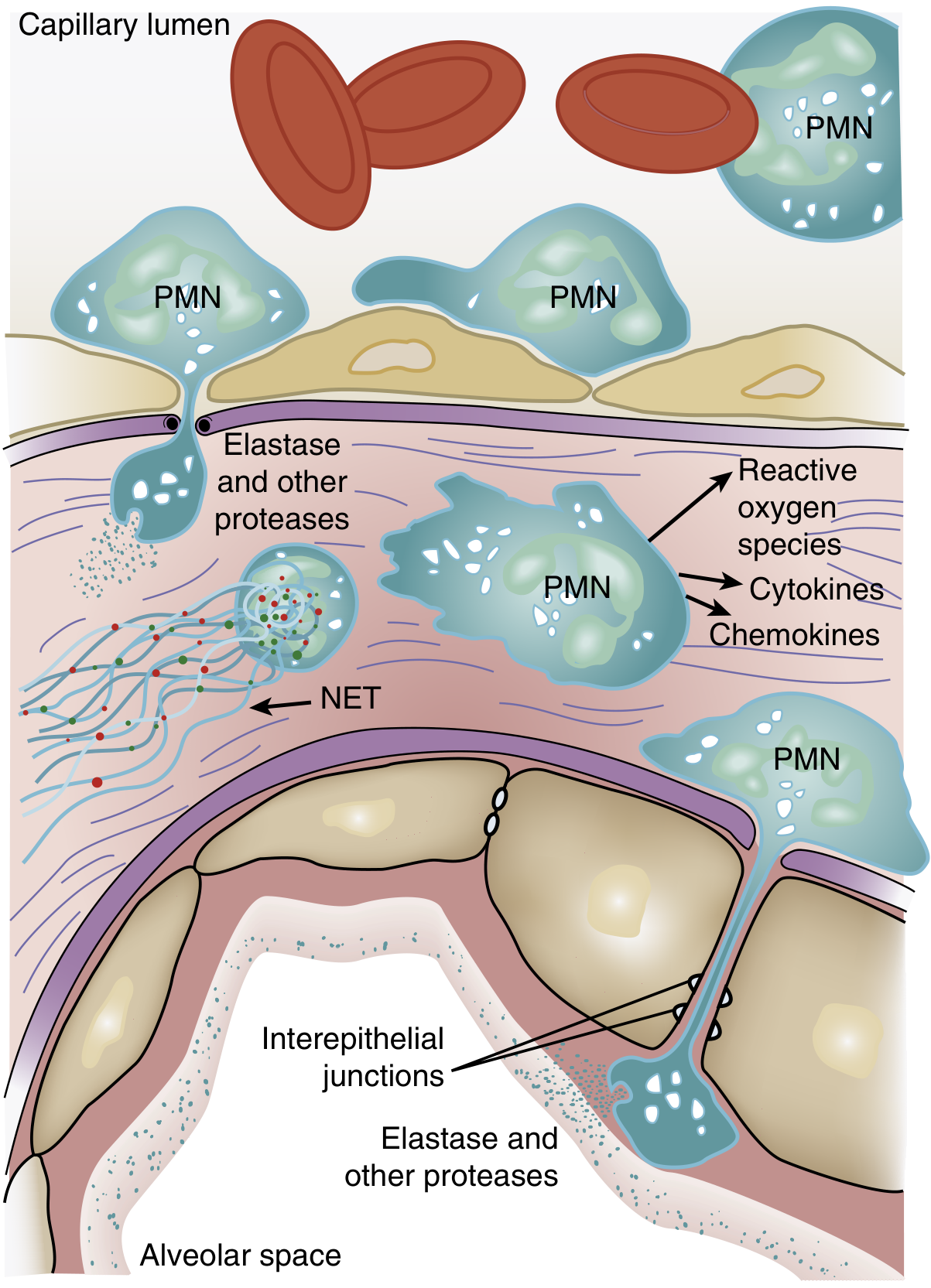
Neutrophil accumulation in the pulmonary microvasculature is a histologic hallmark of ARDS.
Sequence of neutrophil-mediated injury:
-
Sequestration: Activated neutrophils become "stiff" (actin cytoskeleton remodeling) and cannot deform through narrow pulmonary capillaries (diameter ~5-8 µm, smaller than a neutrophil). This causes transient leukopenia - one of the earliest manifestations of ARDS, preceding hypoxemia. Sequestered neutrophils then induce endothelial barrier breakdown, facilitating their own transmigration.
-
Transmigration: PMNs cross the alveolar-capillary membrane, where they release:
- Reactive oxygen species (ROS) - oxidative damage to epithelial and endothelial cells
- Neutrophil elastase (NE) - degrades epithelial/endothelial cadherins (adherens junction components), destroys surfactant protein A, and cleaves growth factors/cytokines
- Matrix metalloproteinases - proteolytic degradation of the extracellular matrix
- TNF-α and IL-1β - amplify the inflammatory cascade
- IL-8 - potent neutrophil chemoattractant, driving further PMN recruitment via PI3K-γ signaling
-
Neutrophil extracellular traps (NETs): Activated neutrophils release web-like structures of DNA, histones, myeloperoxidase, and NE. In ARDS, large quantities of NETs cause direct endothelial damage and thrombus formation. Animal models show that DNase treatment (degrading NETs) attenuates ARDS severity and reduces IL-6 and TNF levels.
Coagulation-inflammation crosstalk: TNF-α stimulates tissue factor expression on endothelium, promotes thrombin/fibrin formation, and inhibits fibrinolysis. Fibrin fragments are themselves chemotactic for neutrophils - creating a positive-feedback loop. Microvascular fibrin deposition worsens gas exchange.
Macrophage/monocyte contribution: Resident alveolar macrophages activated by pattern-recognition receptors (Toll-like receptors) release pro-inflammatory cytokines. Monocyte-derived macrophages and monocyte-platelet aggregates are recruited to the alveolus, further amplifying injury. Even in neutropenic patients with ARDS, alveolar macrophages are likely the alternative source of tissue damage.
Surfactant Dysfunction
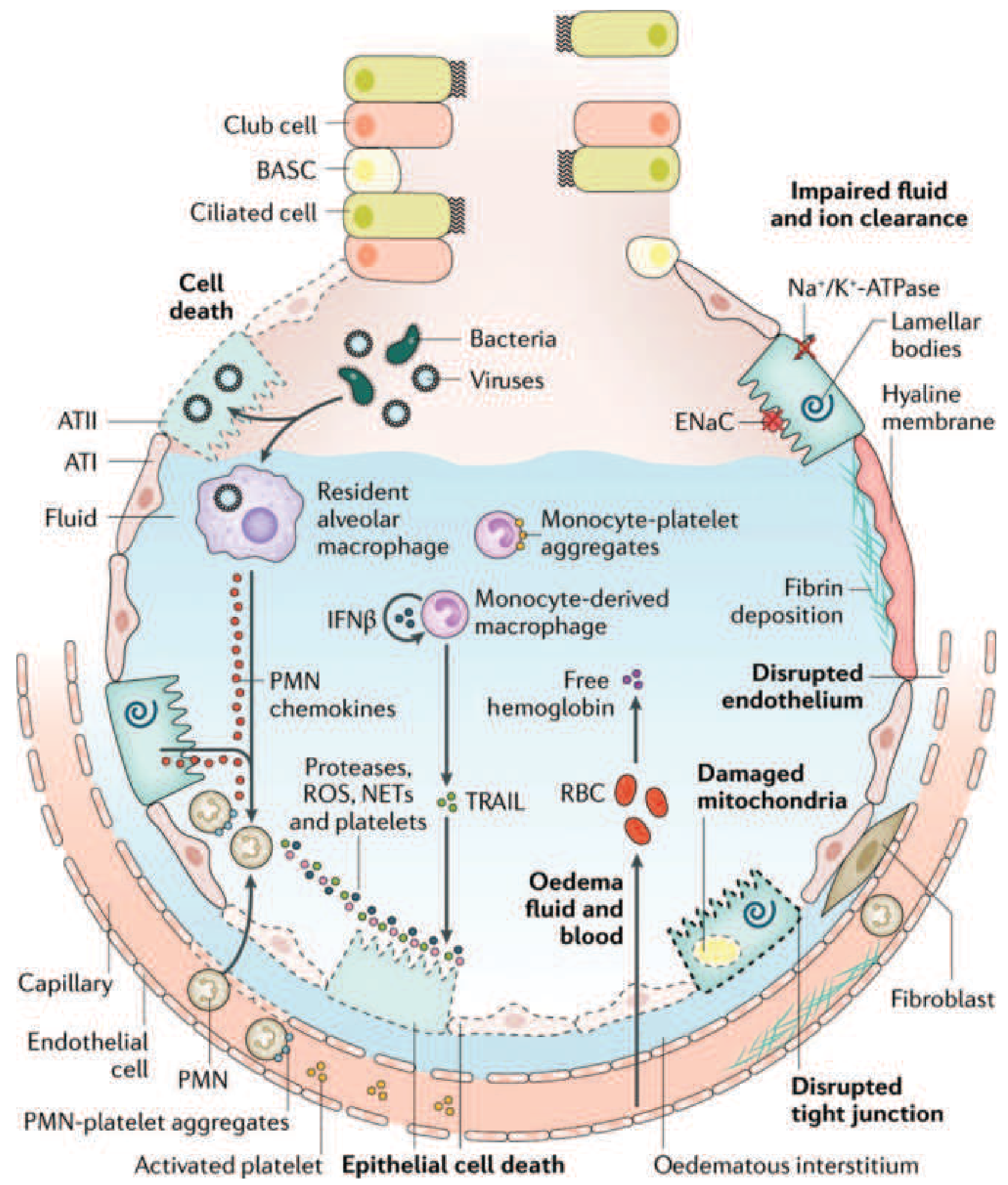
- Damage to Type II pneumocytes reduces surfactant synthesis.
- The composition of surfactant changes: the ratio of dipalmitoylphosphatidylcholine (DPPC) and phosphatidylglycerol is altered, and the proportion of large (active) to small (inactive) surfactant aggregates decreases.
- Plasma proteins leaking into alveoli directly inhibit surfactant function.
- Neutrophil elastase degrades surfactant protein A in vitro, consistent with degraded SP-A found in BAL fluid from ARDS patients.
- Consequence: alveolar units collapse (atelectasis), reducing functional residual capacity (FRC) and worsening ventilation-perfusion mismatch and intrapulmonary shunting.
2. Proliferative Phase (Days 7-21)
Many patients begin recovering during this phase. Key processes:
- Alveolar exudates are organized and cleared.
- Inflammatory infiltrate shifts from neutrophil-predominant to lymphocyte-predominant.
- Type II pneumocytes proliferate along denuded alveolar basement membranes, synthesizing new surfactant and differentiating into Type I cells to restore barrier function.
- Some patients develop early pulmonary fibrosis: elevated N-terminal procollagen peptide III (a marker of collagen synthesis) can be detected in BAL fluid as early as 24 hours after onset, suggesting fibroproliferation may begin in parallel with, not after, the inflammatory phase.
3. Fibrotic Phase (>3 weeks in some patients)
Not all patients progress here. In those who do:
- Alveolar-duct and interstitial fibrosis replaces the earlier inflammatory exudate.
- Emphysema-like changes and large bullae develop from acinar architectural destruction.
- Intimal fibroproliferation in pulmonary microcirculation causes vascular occlusion and pulmonary hypertension.
- Lung compliance decreases, dead space increases, and pneumothorax risk rises.
Ventilator-Induced Lung Injury (VILI) - An Amplifying Mechanism
Once on mechanical ventilation, the injured lung is susceptible to further damage:
- Volutrauma: High tidal volumes cause overdistension of relatively normal alveoli (ARDS is heterogeneous - dependent regions are consolidated/atelectatic, non-dependent regions are relatively normal). Overdistension increases epithelial and endothelial permeability.
- Atelectrauma: Repetitive opening and closing of collapsed terminal lung units generates high shear stresses at the interface of collapsed and aerated tissue.
- Biotrauma: Mechanical stretch activates alveolar macrophages and epithelial cells to release cytokines (TNF-α, IL-1β, IL-6, leukotrienes, prostaglandins, ROS), which can spill into the systemic circulation and contribute to multi-organ dysfunction.
Summary: Pathophysiologic Cascade
Precipitating insult (direct or indirect)
↓
Activation of innate immunity (macrophages, Toll-like receptors, complement)
↓
Cytokine storm (TNF-α, IL-1β, IL-6, IL-8)
↓
Neutrophil sequestration & transmigration into alveoli
↓
Release of proteases (elastase, MMPs), ROS, NETs, cytokines
↓
Disruption of alveolar-capillary barrier
(endothelial tight junction breakdown + Ang2↑ + epithelial death)
↓
Protein-rich edema floods alveoli → hyaline membranes
Surfactant dysfunction → alveolar collapse (atelectasis)
Na+ transport failure → impaired fluid clearance
Coagulation activation → microvascular fibrin thrombi
↓
↓ FRC, ↑ intrapulmonary shunt, V/Q mismatch
↓
Refractory hypoxemia (hallmark of ARDS)
↓
(+/- VILI amplification on mechanical ventilation)
↓
Recovery (proliferative) OR fibrosis progression
Sources: Murray & Nadel's Textbook of Respiratory Medicine; Harrison's Principles of Internal Medicine 22E (2025); Goldman-Cecil Medicine
This is a shared conversation. Sign in to Orris to start your own chat.