Explain Parkinson's disease
Reading File
Finding Sources
Searching PubMed
"Parkinson disease"[MeSH Terms] AND "pathophysiology"
Searching Images
Parkinson disease Lewy body substantia nigra pathology
Reading File
Reading File
Loading Image
Reading File
Finding Sources
Finding Sources
Finding Sources
Reading File
Finding Sources
Reading File
Reading File
Reading File
Loading Image
Loading Image
I now have comprehensive material from multiple authoritative sources. Let me compose the full answer.
Parkinson's Disease
Overview
Parkinson's disease (PD) is a progressive neurodegenerative disorder first described by James Parkinson in 1817 as "involuntary tremulous motion, with lessened muscular power... with a propensity to bend the trunk forward, and to pass from a walking to a running pace, the senses and intellect being uninjured." It is the second most common neurodegenerative disease after Alzheimer's and affects approximately 1% of the population over age 65. Onset typically peaks in the sixth decade; it is uncommon before 30 and slightly more prevalent in men. — Adams and Victor's Principles of Neurology, 12th Ed.
Pathology & Pathophysiology
The hallmark of PD is selective degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNc), with subsequent dopamine depletion in the striatum (nigrostriatal pathway). Grossly, this appears as pallor of the substantia nigra and locus coeruleus due to loss of neuromelanin-containing cells.
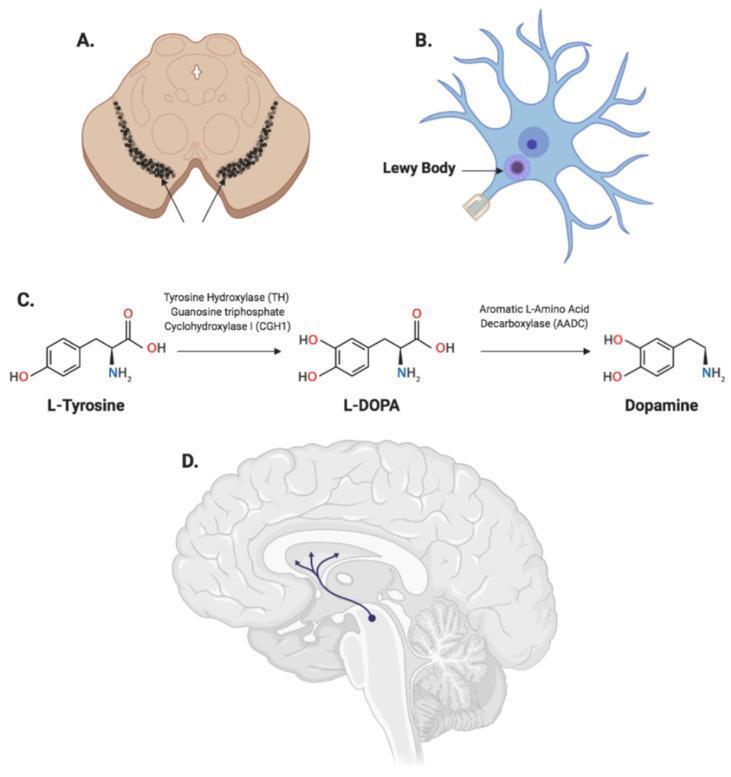
The Lewy Body
Microscopically, surviving neurons contain Lewy bodies — round, eosinophilic cytoplasmic inclusions with a dense pink center and a clear halo. These are composed of aggregated α-synuclein filaments along with neurofilaments and ubiquitin. Dystrophic neurites ("Lewy neurites") also containing α-synuclein are present. — Robbins & Kumar Basic Pathology
α-Synuclein Toxicity
α-Synuclein is a normally soluble presynaptic protein involved in synaptic transmission. In PD, it misfolds and aggregates through a cascade: protofibrils → fibrils → Lewy bodies, causing neurotoxicity. Several mechanisms promote this:
- Gene mutations/duplications in SNCA (α-synuclein gene) increase protein levels or promote oligomerization
- Failure of protein clearance via the ubiquitin-proteasome system (parkin mutations) and autophagy/lysosomal pathways (LRRK2, glucocerebrosidase mutations)
- α-Synuclein–dopamine adduct formation accelerates protofibril accumulation
- Heat shock proteins (HSPs) normally inhibit aggregation but may be overwhelmed
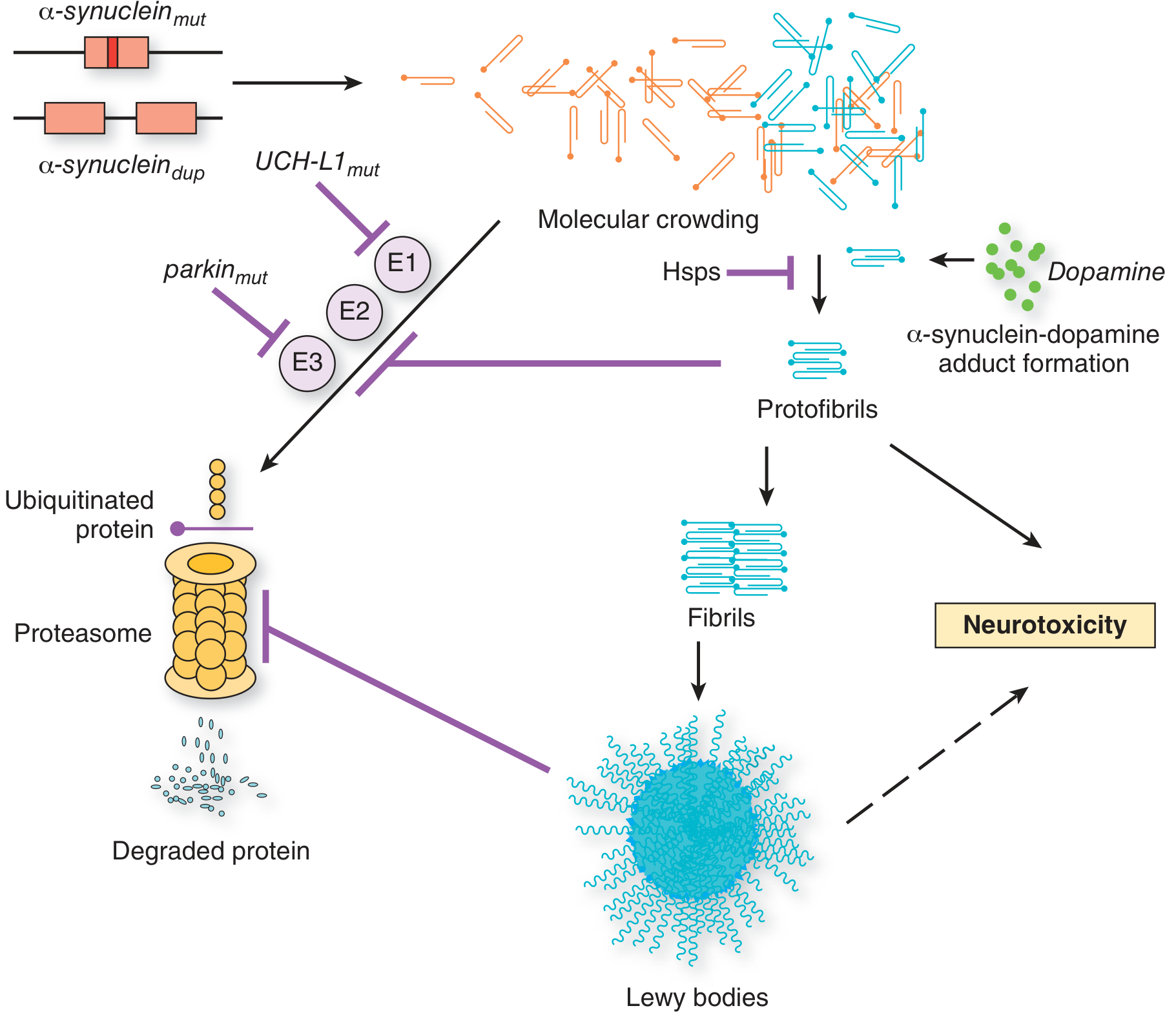
— Adams and Victor's Principles of Neurology, 12th Ed., Fig. 38-6
Neuronal loss beyond the substantia nigra
While the nigrostriatal pathway is most prominent, neuronal loss occurs widely: the locus coeruleus (noradrenergic), raphe nuclei (serotonergic), nucleus basalis of Meynert (cholinergic), dorsal vagal nucleus, and the enteric nervous system — explaining the broad range of non-motor symptoms. — Robbins & Kumar Basic Pathology
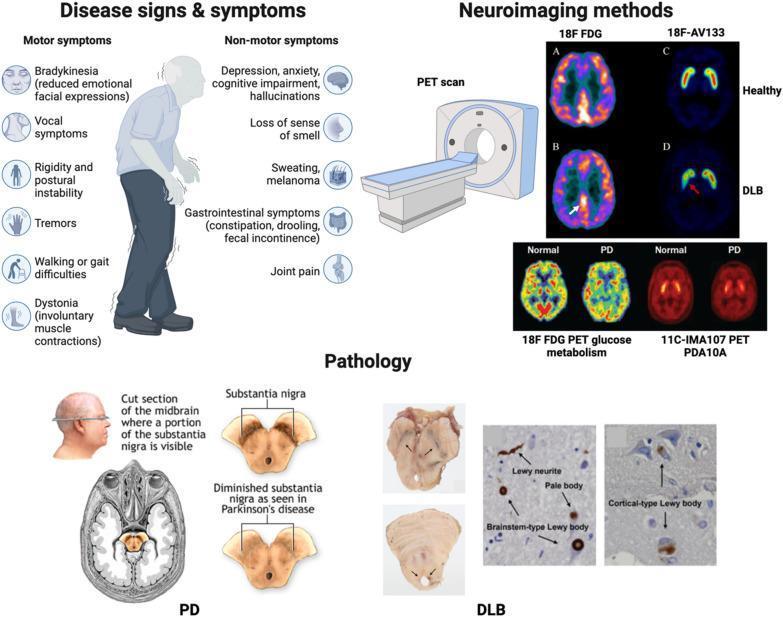
Clinical Features
The classic motor tetrad:
| Feature | Description |
|---|---|
| Tremor | "Pill-rolling" resting tremor, 4–6 Hz; suppressible with voluntary movement; present in ~70% at onset |
| Bradykinesia | Slowness and poverty of movement; expressionless "masked" face; micrographia |
| Rigidity | "Lead-pipe" or "cogwheel" (with superimposed tremor) resistance throughout passive ROM |
| Postural instability | Loss of righting reflexes; festinating gait; retropulsion; falls |
— Adams and Victor's Principles of Neurology (Hoehn & Yahr, 1967)
The disease usually progresses over 10–15 years, eventually producing near-immobility. Death often results from aspiration pneumonia or trauma from falls. — Robbins & Kumar Basic Pathology
Non-Motor Symptoms
These are common, often precede motor symptoms by years, and are frequently under-recognized:
- Neuropsychiatric: Depression (most common psychiatric disturbance, may predate motor onset), anxiety, apathy, psychosis (visual hallucinations in up to 40%, usually medication-induced), impulse control disorders, cognitive impairment, and dementia in advanced disease
- Autonomic: Orthostatic hypotension, constipation, urinary urgency, sweating abnormalities, seborrhea
- Sleep: REM sleep behavior disorder (RBD) — acting out dreams — is a strong prodromal marker; excessive daytime sleepiness
- Sensory: Hyposmia/anosmia (often an early sign), pain
- Gastrointestinal: Dysphagia (subjective in 35%, objective signs in up to 82%), drooling, constipation, esophageal dysmotility
- Neuroimaging: DaTscan (dopamine transporter SPECT) shows reduced striatal dopaminergic uptake
Genetics
Most PD is sporadic, but ~10–15% is familial. Key genetic loci:
| Locus | Gene/Protein | Inheritance | Notes |
|---|---|---|---|
| PARK1/4 | SNCA (α-synuclein) | AD | A53T, A30P mutations; onset 30–40 yr |
| PARK2 | PARK2 (Parkin) | AR | 50% of early-onset inherited PD; no Lewy bodies |
| PARK6 | PINK1 | AR | Mitochondrial kinase |
| PARK7 | PARK7 (DJ-1) | AR | Slow progression; oxidative stress response |
| PARK8 | LRRK2 | AD | Most common autosomal dominant form; common in Ashkenazi Jews |
| — | GBA (glucocerebrosidase) | Risk allele | Lysosomal enzyme; heterozygosity = risk factor |
— Adams and Victor's Principles of Neurology, 12th Ed.
Diagnosis
Diagnosis is clinical, based on the presence of bradykinesia plus at least one of: resting tremor, rigidity, or postural instability, after excluding secondary causes. There is no definitive blood test or imaging biomarker, though DaTscan can support the diagnosis by demonstrating dopamine transporter deficit. The MDS-UPDRS is the standard rating scale for clinical trials.
Treatment
Treatment is symptomatic — no therapy has been proven to halt disease progression.
Pharmacological
| Drug Class | Example | Benefit | Key Side Effects |
|---|---|---|---|
| L-DOPA + carbidopa | Carbidopa/levodopa 25/100 mg | Gold standard; reduces tremor & bradykinesia | Nausea, dyskinesias, orthostatic hypotension, hallucinations |
| Dopamine agonists | Ropinirole, Pramipexole | Moderate motor benefit; reduces L-dopa fluctuations | Sleepiness, impulse control disorders, hallucinations |
| MAO-B inhibitors | Rasagiline, Selegiline | Reduces "off" time; possible neuroprotection | Hypertensive crisis with tyramine/sympathomimetics |
| COMT inhibitors | Entacapone | Prolongs L-dopa effect | Diarrhea, dyskinesias, urine discoloration |
| Glutamate antagonist | Amantadine | Smooths motor fluctuations; reduces dyskinesias | Leg edema, confusion, insomnia |
| Anticholinergics | Benztropine, Trihexyphenidyl | Tremor reduction (mainly) | Dry mouth, urinary retention, confusion |
Over time, L-DOPA becomes less effective and causes motor fluctuations ("on-off" phenomena) and dyskinesias due to denervation hypersensitivity of striatal dopamine receptors. — Adams and Victor's Principles of Neurology
Surgical
Deep brain stimulation (DBS) — electrodes implanted in the globus pallidus interna (GPi) or subthalamic nucleus (STN) — modulates basal ganglia circuitry and allows significant reduction in L-DOPA dose in selected patients. The current disease model holds that dopamine loss leads to dysfunction of the cortico-striato-thalamo-cortical circuit, resulting in deficient thalamo-cortical drive and impaired motor cortex facilitation (bradykinesia). — Bradley and Daroff's Neurology, 8th Ed.
Non-pharmacological
- Physiotherapy (gait, balance, fall prevention)
- Speech therapy (voice volume — Lee Silverman Voice Treatment; dysphagia management)
- Occupational therapy
- Exercise — particularly rhythmic aerobic exercise, tai chi, and boxing programs — has Level B evidence for improving motor function
Differential Diagnosis (Secondary Parkinsonism)
- Drug-induced parkinsonism (dopamine antagonists: haloperidol, metoclopramide, prochlorperazine)
- Vascular parkinsonism ("lower body parkinsonism")
- Multiple system atrophy (MSA) — autonomic failure + parkinsonism ± cerebellar signs
- Progressive supranuclear palsy (PSP) — vertical gaze palsy, early falls, no tremor
- Dementia with Lewy bodies (DLB) — dementia precedes or co-presents with parkinsonism; prominent hallucinations; neuroleptic sensitivity
- Corticobasal degeneration (CBD) — asymmetric apraxia, alien limb
Sources: Robbins & Kumar Basic Pathology; Adams and Victor's Principles of Neurology, 12th Ed.; Bradley and Daroff's Neurology in Clinical Practice, 8th Ed.; Sleisenger and Fordtran's Gastrointestinal and Liver Disease.
This is a shared conversation. Sign in to Orris to start your own chat.