Crao
Checking Date & Time
Reading File
Finding Sources
Searching PubMed
"central retinal artery occlusion" AND management
Reading File
Reading File
Searching Images
central retinal artery occlusion fundus
Reading File
Reading File
Reading File
Reading File
Loading Image
Loading Image
Here is a comprehensive overview of Central Retinal Artery Occlusion (CRAO):
Central Retinal Artery Occlusion (CRAO)
Pathophysiology
The ophthalmic artery (first branch of the internal carotid artery) gives rise to the central retinal artery, which supplies the inner retina. Occlusion causes infarction of the inner retina → ischemic opacification and edema. The foveola is the thinnest retinal area, allowing the intact underlying choroidal circulation to remain visible — creating the pathognomonic "cherry-red spot".
Fundus Appearance
Classic CRAO (Tintinalli's Emergency Medicine):
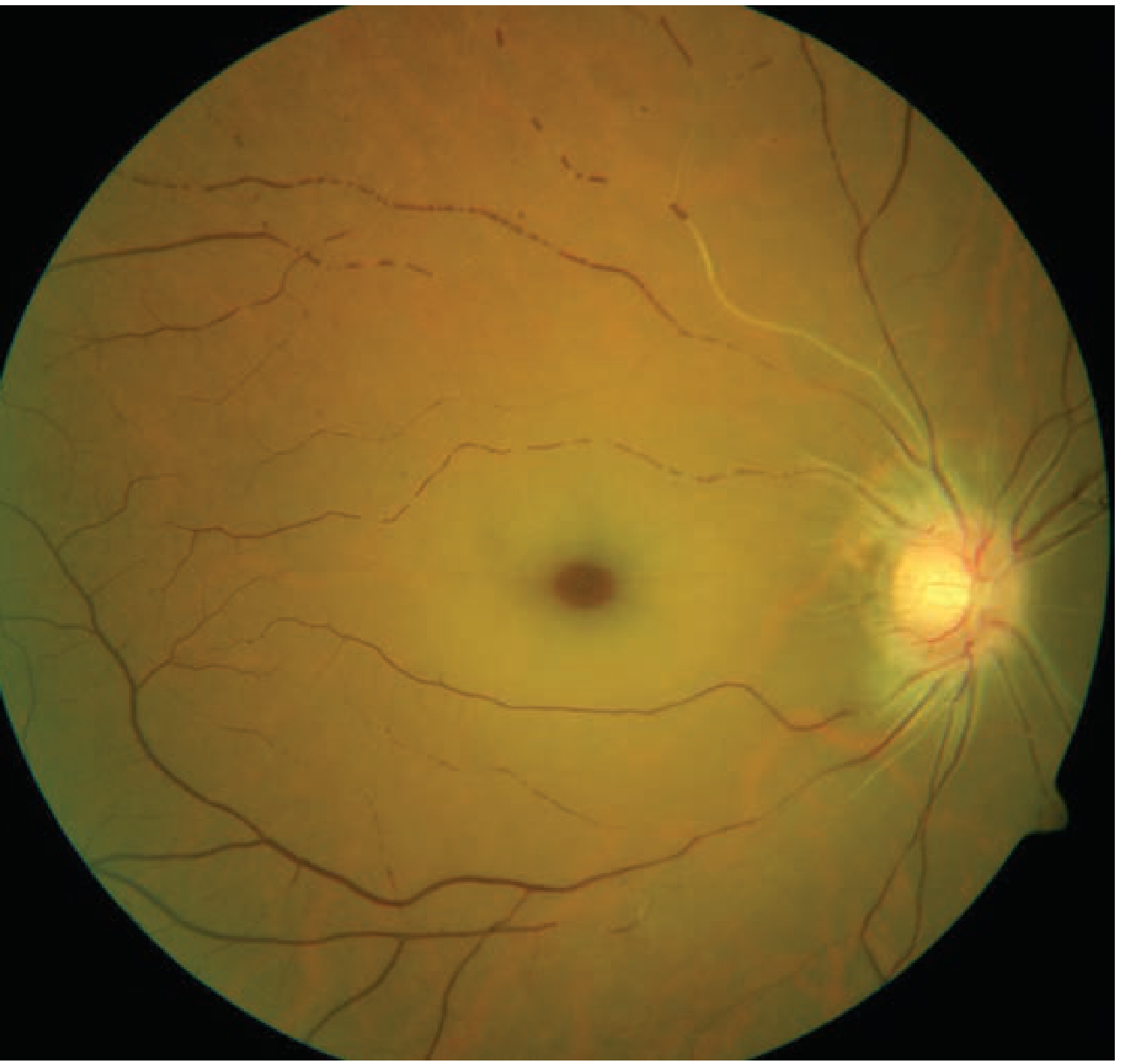
Diffuse retinal pallor with macular cherry-red spot. Note the attenuated arterioles.
CRAO vs BRAO comparison:
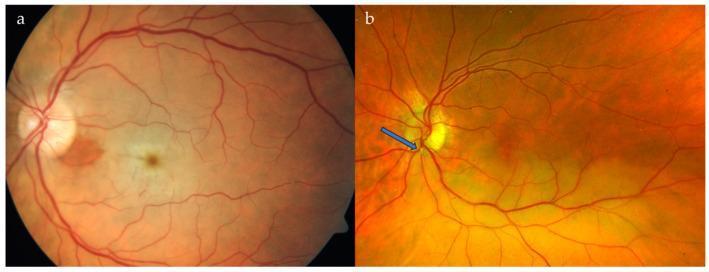
(a) CRAO: generalized retinal pallor with cherry-red spot. (b) BRAO: localized whitening with visible embolus (blue arrow).
Symptoms
- Sudden, profound, painless, monocular vision loss (over seconds)
- VA is severely reduced — counting fingers to light perception in ~94% of eyes
- Often preceded by episodes of amaurosis fugax
- Pain suggests giant cell arteritis (GCA)
- Absence of light perception → suspect GCA or ophthalmic artery occlusion (not just CRAO)
— Kanski's Clinical Ophthalmology, p. 552; Wills Eye Manual, p. 782
Signs
| Finding | Detail |
|---|---|
| Cherry-red spot | Pathognomonic; may be subtle early |
| Retinal pallor/opacification | Posterior pole; may take hours to develop |
| Profound RAPD | Sometimes amaurotic pupil |
| Arteriolar attenuation | Narrowed retinal vessels |
| Boxcarring / segmentation | Blood column segmentation in arterioles |
| Emboli | Visible in ~20% of cases |
| Peripapillary swelling | Optic disc may be swollen |
Cilioretinal artery sparing: Present in ~1/3 of eyes; if the fovea is supplied by a cilioretinal artery, central vision may be preserved despite CRAO.
Late changes: Retinal cloudiness and cherry-red spot disappear over days-weeks → optic atrophy, vessel sheathing, inner retinal atrophy.
Complications: ~2% develop retinal/disc neovascularization; rubeosis iridis in up to ~20% (typically at 4–5 weeks).
— Kanski's Clinical Ophthalmology, pp. 551–552; Wills Eye Manual, p. 782
Etiology
| Category | Examples |
|---|---|
| Embolism | Cholesterol (Hollenhorst plaques — from carotid atheromas), calcium (from cardiac valves), platelet-fibrin |
| Thrombosis | In situ thrombosis |
| GCA | Arteritic CRAO — always rule out |
| Collagen vascular disease | SLE, polyarteritis nodosa |
| Hypercoagulable states | Antiphospholipid syndrome, factor V Leiden, protein C/S deficiency, polycythemia |
| Other | Elevated IOP (glaucoma), migraine/vasospasm, trauma, sickle cell, syphilis, Behçet disease |
— Wills Eye Manual, pp. 783–784
Workup
Treat as an acute stroke. AAO 2018 guidelines: send immediately to an emergency department with a stroke center.
- ESR, CRP, platelets — rule out GCA in patients ≥55 years (if no visible embolus)
- Blood pressure check
- Labs: Fasting glucose, HbA1c, CBC with differential, PT/PTT. In patients <50 years or with atypical features: lipid panel, ANA, RF, syphilis serology (RPR/VDRL + FTA-ABS), SPEP, hypercoagulable workup
- Carotid duplex Doppler US
- Cardiac workup: ECG, echocardiography, Holter or bubble study
- OCT (confirms diagnosis; highly reflective embolic plaque in superficial nerve head on OCT); consider IVFA or ERG if diagnosis uncertain
Treatment
Window: Irreversible retinal damage is thought to occur after ~4–6 hours of ischemia. Treatment should ideally be within 90–120 minutes of onset.
No treatment has been proven effective in RCTs. All ocular measures are anecdotal.
Systemic / Stroke pathway
- Immediate transfer to stroke centre
- IV tPA within 4.5 hours of onset has some supporting evidence (multiple observational studies)
- If GCA suspected: high-dose systemic corticosteroids immediately
- Cardiology and neurology consultation
Ocular measures (attempted within 90–120 min, limited evidence)
| Measure | Rationale |
|---|---|
| Ocular massage (digital or three-mirror contact lens) | Mechanical dislodgement of embolus/thrombus; alternating pressure 10–15 s × 3–5 min |
| Anterior chamber paracentesis (27G needle, 0.1–0.2 mL aqueous removed) | Acute IOP reduction → arterial reperfusion |
| IOP-lowering agents | Topical timolol 0.5%, apraclonidine 1%; IV acetazolamide 500 mg |
| Carbogen inhalation (95% O₂ + 5% CO₂) or paper bag rebreathing | Vasodilation + retard ischemia; evidence very limited |
| Nd:YAG laser embolysis | If embolus visible; limited experience |
— Kanski's Clinical Ophthalmology, p. 553–554; Wills Eye Manual, p. 785; Tintinalli's Emergency Medicine, p. 1597
Differential Diagnosis of Cherry-Red Spot
| Condition | Key differentiator |
|---|---|
| Ophthalmic artery occlusion | No cherry-red spot; entire retina white; worse prognosis |
| Commotio retinae (Berlin edema) | History of blunt trauma; resolves spontaneously |
| Tay-Sachs / Niemann-Pick A | Bilateral; presents in infancy with systemic features |
Follow-Up
- Repeat eye exam 1–4 weeks post-event
- Monitor for neovascularization of iris (NVI), disc (NVD), angle (NVA), or retina (NVE) — occurs in up to 20% at ~4 weeks
- If neovascularization develops: panretinal photocoagulation (PRP) and/or intravitreal anti-VEGF
- Long-term systemic cardiovascular risk factor management
Prognosis
Poor overall. In two-thirds of cases final VA is worse than 6/120 (20/400); only ~1 in 5 patients achieves VA ≥ 6/12 (20/40).
Recent evidence (PMID 41006752): A 2025 systematic review and meta-analysis confirmed that retinal artery occlusion patients carry a significantly elevated risk of concurrent and subsequent ischemic cerebrovascular disease — reinforcing the importance of the acute stroke workup.
— Kanski's Clinical Ophthalmology, p. 554; Wills Eye Manual, p. 785; Tintinalli's Emergency Medicine, p. 1597
Retinitis pigmentosa for 20 marks
Checking Date & Time
Finding Sources
Reading File
Reading File
Reading File
Reading File
Searching Images
retinitis pigmentosa bone spicule fundus
Searching PubMed
"retinitis pigmentosa" AND treatment
Loading Image
Loading Image
Reading File
Reading File
Retinitis Pigmentosa (RP)
Definition
Retinitis pigmentosa (RP), or pigmented retinal degeneration, is a clinically and genetically diverse group of inherited diffuse retinal degenerative diseases that initially predominantly affect rod photoreceptors, with subsequent degeneration of cones and the retinal pigment epithelium (RPE). Despite the name, RP is not an inflammatory process.
— Kanski's Clinical Ophthalmology, p. 646
Epidemiology
- Most common hereditary retinal degeneration
- Prevalence: 1 in 3,000 to 1 in 5,000
- No sex predilection; affects all races
- Onset, rate of progression, and severity are related to the mode of inheritance
Genetics & Inheritance
| Inheritance Pattern | Frequency | Key Features |
|---|---|---|
| Autosomal recessive (AR) | Most common | Severe, early onset (childhood/young adult); intermediate severity |
| Autosomal dominant (AD) | Second most common | Least severe; gradual adult onset; variable penetrance; late cataract |
| X-linked recessive (XLR) | Rarest | Most severe; reduction to 6/60 or less by 5th decade; female carriers show "tapetal reflex" or salt-and-pepper fundus |
| Sporadic | ~40–50% | May represent any of the above without family history (incomplete penetrance, small families, etc.) |
Genetic basis: Mutations in >100 gene loci cause non-syndromic RP. Key mechanisms:
- Phototransduction cascade — most AD cases: rhodopsin gene mutations
- Retinoid cycle — RPE65 mutations (target of gene therapy)
- Photoreceptor structure — peripherin (PRPH2) mutations
- Connecting cilium — RPGR gene mutations (90% of X-linked cases)
— Kanski's Clinical Ophthalmology, p. 646; Harrison's Principles of Internal Medicine 22e, p. 275
Pathophysiology
- Primary degeneration of rod photoreceptors → night blindness and peripheral field loss
- Secondary degeneration of cones → central vision loss (late)
- Degeneration of RPE → accumulation of lipofuscin, migration of RPE cells into the retina forming bone-spicule deposits around perivascular spaces
- Inner retina remains relatively intact initially, preserving the ERG b-wave only transiently
Clinical Features
Symptoms (in order of progression)
- Nyctalopia (night blindness) — often the presenting symptom; earliest manifestation of rod dysfunction
- Peripheral visual field loss — gradual constriction; "tunnel vision"
- Ring scotoma — characteristic progressive visual field defect (mid-peripheral ring that expands inward and outward)
- Photopsia — flickering/flashing lights
- Loss of central vision — late feature (unless early cataract develops)
- Colour vision loss — variable; later in disease
Signs — Classic Triad
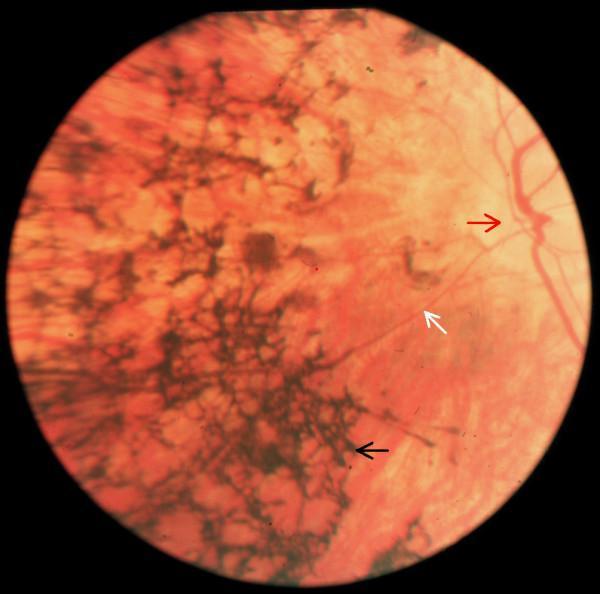
Advanced RP: extensive bone-spicule pigmentation, severely attenuated retinal vasculature, and waxy disc pallor.
| Sign | Description |
|---|---|
| Bone-spicule pigmentation | Dark, irregular, branching intraretinal perivascular pigment deposits; initially in mid-periphery; spreads anteriorly and posteriorly |
| Arteriolar attenuation | Marked narrowing of retinal blood vessels; "thread-like" vessels |
| Waxy disc pallor | Yellowish-waxy optic disc pallor due to secondary optic atrophy |
Additional signs:
- Vitreous cells — most consistent sign (Wills Eye)
- Posterior subcapsular cataract (PSC) — common in all forms
- Cystoid macular oedema (CME) — in ~15% on OCT
- Epiretinal membrane (ERM)
- Optic disc drusen — more frequent than in general population
- Tessellated fundus (unmasking of choroidal vessels due to RPE atrophy)
Note: RP sine pigmento — every patient passes through this phase early; bone spicules develop after photoreceptor degeneration, so absence of pigment does not exclude diagnosis.
— Kanski's Clinical Ophthalmology, pp. 646–647; Wills Eye Manual, p. 878
Natural History & Progression
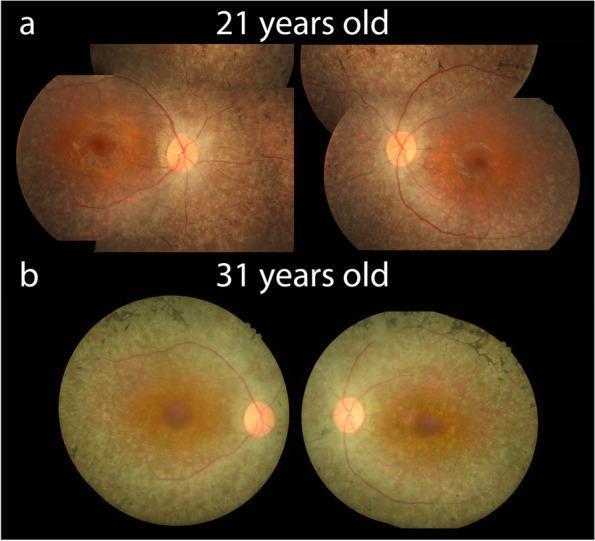
Bilateral RP progression over 10 years — (a) age 21: mild changes; (b) age 31: waxy disc pallor, severe vessel attenuation, and widespread RPE atrophy.
- Perimetry initially shows small mid-peripheral scotomas → coalesce into ring scotoma → tunnel vision → extinction
- XLR: central VA reduced to ≤6/60 by the 5th decade
- AD: best prognosis; AR: intermediate severity
Investigations
| Investigation | Findings |
|---|---|
| Electroretinogram (ERG) (gold standard) | Early: reduced scotopic (rod) responses. Later: reduced photopic (cone) responses. Terminal: extinguished ERG. Distinguishes RP from optic nerve disease. |
| Fundus Autofluorescence (FAF) | Abnormal perimacular hyperautofluorescent ring (RPE lipofuscin); patchy peripheral hypo-AF. Distinguishes RP from normal in 95% of cases. FAF also shows centrifugal hyperAF lines in X-linked carrier females. |
| OCT | Macular oedema (~15%), macular atrophy (~40%); prognostic value; detects ERM |
| Perimetry (visual field testing) | Mid-peripheral ring scotoma → concentric constriction |
| Dark adaptation testing | Prolonged; useful in equivocal early cases |
| Fluorescein angiography (FA) | Variable RPE window defects, CME may be seen |
| Genetic analysis | Identifies specific mutations; guides genetic counselling and trial eligibility |
| Serology | Rule out infectious mimics (e.g., syphilis) where relevant |
— Kanski's Clinical Ophthalmology, p. 647
Differential Diagnosis — Conditions Mimicking RP
| Condition | Distinguishing Feature |
|---|---|
| Syphilitic chorioretinitis | Serology positive; treatable |
| Chloroquine / hydroxychloroquine toxicity | Drug history; bull's-eye maculopathy |
| Thioridazine (phenothiazine) toxicity | Drug history |
| Old rubella retinopathy | Salt-and-pepper fundus; history |
| Resolved exudative retinal detachment | History; demarcation line |
| Choroideremia | X-linked; scalloped chorioretinal atrophy; affects males severely |
| Gyrate atrophy | Elevated plasma ornithine; AD |
| Kearns-Sayre syndrome | Salt-and-pepper fundus; CPEO; ptosis; cardiac conduction defects |
Complications
- Posterior subcapsular cataract (most common)
- Cystoid macular oedema (CME) — reversible with carbonic anhydrase inhibitors
- Epiretinal membrane
- Open-angle glaucoma (~3%)
- Posterior vitreous detachment
- Neovascularization (rare)
Associated Systemic Syndromes (Syndromic RP)
20–30% of RP cases are associated with systemic disease (usually AR or mitochondrial):
| Syndrome | Key Features |
|---|---|
| Usher syndrome (AR) | RP + sensorineural deafness ± vestibular dysfunction. Most common cause of combined blindness+deafness. Type I (MYO7A): profound congenital deafness + severe RP; Type III: progressive hearing loss. Cochlear implant for deafness. |
| Bassen-Kornzweig (Abetalipoproteinemia) (AR) | Failure to absorb fat-soluble vitamins (A, D, E, K) → spinocerebellar ataxia, acanthocytosis (thorny RBCs), RP-like retinopathy. Treat with vitamin supplementation + low-fat diet. |
| Refsum disease (AR) | Phytanic acid accumulation → ichthyosis, peripheral neuropathy, cerebellar ataxia, deafness, RP. Treat with low-phytanic acid diet. |
| Bardet-Biedl syndrome (AR) | RP/bull's-eye maculopathy + polydactyly + obesity + renal dysfunction + mental handicap. 80% have severe visual changes by age 20. |
| Kearns-Sayre syndrome (mitochondrial) | Salt-and-pepper retinopathy + CPEO + bilateral ptosis + cardiac conduction defects. Single mitochondrial DNA deletion. |
| Leber congenital amaurosis (AR) | Severe early-onset rod-cone dystrophy (birth–infancy); nystagmus, poor fixation, reduced/absent ERG. RPE65 mutations targetable by gene therapy (Luxturna). |
| Neuropathy, Ataxia, RP (NARP) | Mitochondrial; peripheral neuropathy + cerebellar ataxia + RP |
Management
Currently Available
| Intervention | Indication / Evidence |
|---|---|
| Annual ophthalmology follow-up | Detect treatable complications; monitor progression |
| Cataract surgery | Often substantially improves vision |
| Carbonic anhydrase inhibitors (oral/topical acetazolamide) | CME — anecdotal/limited RCT evidence |
| Anti-VEGF agents | Adjunct for CME |
| Low-vision aids & rehabilitation | When central vision is reduced |
| Vitamin A palmitate (15,000 IU/day) + lutein (12 mg/day) | Some evidence of slowing mid-peripheral field loss in non-smokers (avoid in pregnancy; monitor LFTs and vitamin A levels) |
| Smoking cessation | Advised in all patients |
| Genetic counselling | Based on identified mutation and inheritance pattern |
Emerging / Approved Therapies
| Therapy | Detail |
|---|---|
| Luxturna (voretigene neparvovec) | FDA-approved gene therapy for RPE65 mutation-associated RP/Leber congenital amaurosis; subretinal injection |
| Retinal implants (epiretinal / subretinal microchips) | Used in advanced end-stage RP; some improvement in light perception |
| Gene therapy trials | Ongoing for RPGR (X-linked), CNGB1, and other mutations |
| Optogenetics | Research phase; converts surviving retinal cells into light-sensitive cells |
— Kanski's Clinical Ophthalmology, p. 647; Wills Eye Manual, p. 879
Prognosis
- No cure currently; course is progressive and irreversible
- Rate of progression highly variable depending on genetic mutation and inheritance pattern
- AD: best prognosis (slow progression, later onset)
- XLR: worst prognosis (most severe, rapid progression)
- Patients often retain some useful central vision into middle age
- Regular follow-up critical to manage complications and access emerging therapies
Summary Box — Exam Points
| Feature | Key Fact |
|---|---|
| Definition | Inherited rod-cone dystrophy; rods affected first |
| Prevalence | 1:3,000–5,000; most common hereditary retinal degeneration |
| Classic triad | Bone spicules + arteriolar attenuation + waxy disc pallor |
| First symptom | Nyctalopia (night blindness) |
| Visual field defect | Ring scotoma → tunnel vision |
| Most sensitive test | Full-field ERG |
| Most consistent sign | Vitreous cells |
| Gene therapy | Luxturna for RPE65 mutations |
| Most common complication | Posterior subcapsular cataract |
| Most common syndrome | Usher syndrome (RP + deafness) |
| Severity order | AD (mildest) < AR < XLR (most severe) |
— Kanski's Clinical Ophthalmology 10e; Wills Eye Manual; Harrison's Principles 22e
Give this for 10 marks
Based on the comprehensive content already in context, here is a concise 10-mark answer on Retinitis Pigmentosa:
Retinitis Pigmentosa
Definition
RP is the most common inherited retinal degeneration (prevalence 1:3,000–5,000), characterized by progressive degeneration of rod photoreceptors first, followed by cones and RPE. It is a non-inflammatory condition despite its name.
Inheritance
| Pattern | Severity |
|---|---|
| Autosomal Dominant | Mildest; late onset |
| Autosomal Recessive | Intermediate |
| X-linked Recessive | Most severe; near blindness by 5th decade |
| Sporadic | Any of the above without family history |
Key genes: rhodopsin (AD), RPGR (90% of X-linked), RPE65 (target of gene therapy).
Clinical Features
Symptoms (in order of onset):
- Nyctalopia (night blindness) — earliest symptom; rod dysfunction
- Peripheral visual field loss → ring scotoma → tunnel vision
- Photopsia
- Central vision loss — late
Classic Triad on Fundoscopy:
- Bone-spicule pigmentation — dark, branching perivascular deposits in mid-periphery
- Arteriolar attenuation — thread-like narrowed vessels
- Waxy disc pallor — secondary optic atrophy
Other signs: Vitreous cells (most consistent), posterior subcapsular cataract (most common complication), CME, ERM.
Investigations
- ERG (gold standard) — reduced scotopic → photopic → extinguished
- FAF — perimacular hyperautofluorescent ring; distinguishes RP in 95%
- OCT — detects CME (~15%), macular atrophy (~40%)
- Perimetry — ring scotoma → concentric constriction
- Genetic analysis — counselling and trial eligibility
Associated Syndromes
| Syndrome | Association |
|---|---|
| Usher syndrome | RP + sensorineural deafness (most common) |
| Refsum disease | RP + ichthyosis + ataxia + neuropathy |
| Bardet-Biedl | RP + polydactyly + obesity + renal dysfunction |
| Bassen-Kornzweig | RP + acanthocytosis + spinocerebellar ataxia |
| Kearns-Sayre | Salt-and-pepper retinopathy + CPEO + cardiac defects |
Management
- Luxturna — FDA-approved gene therapy for RPE65 mutations
- Vitamin A palmitate 15,000 IU/day + lutein 12 mg/day (may slow field loss)
- Cataract surgery when indicated
- Carbonic anhydrase inhibitors for CME
- Low vision aids and rehabilitation
- Annual follow-up; genetic counselling; smoking cessation
Prognosis
No cure; progressive and irreversible. AD has the best prognosis; X-linked the worst. Central vision often preserved into middle age.
This is a shared conversation. Sign in to Orris to start your own chat.