Tell me about colorectal cancer
Reading File
Finding Sources
Searching PubMed
"colorectal cancer"[MeSH Terms] AND ("diagnosis" OR "treatment")
Searching the Web
colorectal cancer overview epidemiology diagnosis treatment 2025
Reading File
Reading File
Finding Sources
Searching Images
colorectal cancer pathology staging tumor
Reading File
Reading File
Reading File
Reading File
Reading File
Reading File
I have comprehensive content from multiple authoritative textbooks. Now I'll compile a thorough, well-organized response.
Colorectal Cancer (CRC)
Epidemiology
Colorectal cancer is the third most common cancer worldwide and the second leading cause of cancer-related death globally. In the United States, it accounts for approximately 7.5% of all new cancer cases, with an estimated 158,850 new cases and 55,230 deaths projected for 2026 (SEER data). Roughly 6% of Americans will develop CRC in their lifetime, with an overall 5-year survival rate of approximately 62%.
Incidence has been declining in adults over 50 — from 60.5 per 100,000 in 1976 to ~40.7 per 100,000 in 2009–2013 — largely due to screening and improved therapies. However, rates in patients younger than 50 are rising, with significant increases expected in the 20–34 age group by 2030, for reasons that remain unclear.
- Age distribution: Incidence starts rising at age 40, doubling with each decade thereafter
- Sex: Right colon cancer is more common in women; rectal cancer is more common in men
- Tietz Textbook of Laboratory Medicine, 7th Ed.; Swanson's Family Medicine Review
Anatomic Distribution
| Location | % of All CRC |
|---|---|
| Rectum | 30% |
| Sigmoid colon | 20% |
| Ascending colon | 25% |
| Descending colon | 15% |
| Transverse colon | 10% |
Swanson's Family Medicine Review
Risk Factors
Dietary/lifestyle:
- High intake of red and processed meats (WHO classifies processed meat as a carcinogen, red meat as a probable carcinogen)
- Obesity and sedentary lifestyle
- Alcohol consumption and cigarette smoking
- Low fiber, low fruit/vegetable diet
- High-fat diet
Medical conditions:
- Ulcerative colitis and Crohn's disease
- Colorectal polyps (adenomas)
- Prior radiation exposure
Genetic/familial (lifetime CRC risk by family history):
- No affected first-degree relative: ~4%
- One first-degree relative: ~9%
- More than one first-degree relative: ~16%
- First-degree relative diagnosed before age 45: ~15%
Goldman-Cecil Medicine; Swanson's Family Medicine Review
Pathogenesis & Molecular Biology
CRC is a heterogeneous, multipathway disease. The classical model is the adenoma-carcinoma sequence (Fearon & Vogelstein, 1990): normal mucosa → hyperplasia → adenoma → carcinoma. Tubular adenomas carry ~22% malignant risk; villous adenomas carry ~40%.
Sporadic CRC Subtypes
-
Chromosomally unstable (CIN) CRC — most common, involves sequential mutations in tumor suppressor genes (TSGs) and oncogenes:
- APC gene loss → early adenoma formation
- KRAS oncogene mutation (codons 12, 13, 61) — present in 35–45% (some series up to 75%) of CRCs; drives constitutive RAS/RAF/ERK signaling, promoting proliferation, invasion, angiogenesis, and metastasis
- SMAD-2/SMAD-4, DCC, TP53 loss → progression to carcinoma
-
Microsatellite unstable (MSI) CRC — defective DNA mismatch repair (MMR); accounts for ~15% of sporadic CRCs. Also the hallmark of Lynch syndrome.
-
CpG island methylator phenotype (CIMP) — epigenetic silencing of key genes (e.g., MLH1 methylation); associated with BRAF mutations and serrated pathway cancers.
Key Molecular Markers
- EGFR: Overexpressed in up to 80% of CRCs; high expression → poor prognosis. Targeted by anti-EGFR monoclonal antibodies (cetuximab, panitumumab) — but only effective in KRAS/RAS wild-type tumors
- KRAS: Mutation = primary predictor of anti-EGFR therapy resistance; testing mandatory before cetuximab/panitumumab
- BRAF V600E: Found in ~15% of unselected CRCs and up to 70% of serrated polyposis syndrome cancers; associated with poor prognosis in MSS tumors
- MSI/MMR status: Predicts benefit from immunotherapy (pembrolizumab, nivolumab) in MSI-High tumors
Henry's Clinical Diagnosis and Management by Laboratory Methods
Hereditary Syndromes
| Syndrome | Gene(s) | Key Features | CRC Risk |
|---|---|---|---|
| Familial Adenomatous Polyposis (FAP) | APC | Hundreds to thousands of colonic polyps; autosomal dominant | Near 100% by age 40 |
| Lynch Syndrome (HNPCC) | MLH1, MSH2, MSH6, PMS2 | Defective MMR; right colon predominance; autosomal dominant | 50–80% lifetime |
| Peutz-Jeghers Syndrome (PJS) | STK11/LKB1 | Hamartomatous polyps, mucocutaneous pigmentation; autosomal dominant | ~39% by age 65 |
| Serrated Polyposis Syndrome | MUTYH (some cases) | ≥100 hyperplastic/serrated polyps; BRAF mutations common | 37–69% |
| MYH (MUTYH)-Associated Polyposis | MUTYH | Autosomal recessive; adenomas without APC mutation | 177-fold ↑ with biallelic mutations |
Henry's Clinical Diagnosis and Management by Laboratory Methods
Clinical Presentation
Symptoms vary by tumor location:
- Right colon (ascending): Often insidious — iron-deficiency anemia, fatigue, occult blood; rarely obstructs due to liquid stool
- Left colon / sigmoid: Change in bowel habits, pencil-thin stools, hematochezia, obstructive symptoms
- Rectum: Tenesmus, fresh rectal bleeding, sense of incomplete evacuation
Early CRC is often asymptomatic — the rationale for screening programs.
Common symptoms at presentation include: altered bowel habits, weight loss, anemia, and positive fecal occult blood.
Diagnosis & Staging
Diagnostic Tools
- Colonoscopy: Gold standard — allows visualization, biopsy, and polypectomy. Preferred when resources allow.
- CT colonography (virtual colonoscopy): Non-invasive; recommended every 5 years
- Flexible sigmoidoscopy: Every 5 years (or 10 years with annual FOBT)
- Biopsy and histopathology: Required for diagnosis; ~95% are adenocarcinomas
Tumor Markers
- CEA (carcinoembryonic antigen): Most clinically relevant serum marker. Used primarily for:
- Monitoring response to treatment
- Detecting recurrence after surgery
- NOT recommended for primary screening (lacks sensitivity/specificity)
- Fecal immunochemical test (FIT): Detects globin component of hemoglobin; better specificity and automation than older guaiac-based tests; preferred for population-based screening
- Multi-target stool DNA test (Cologuard®): FDA-approved; detects methylated BMP3 and NDRG4, mutant KRAS, beta-actin, and hemoglobin; superior sensitivity to FIT for cancer and advanced precancerous lesions, but less specific
Tietz Textbook of Laboratory Medicine
TNM Staging (AJCC)
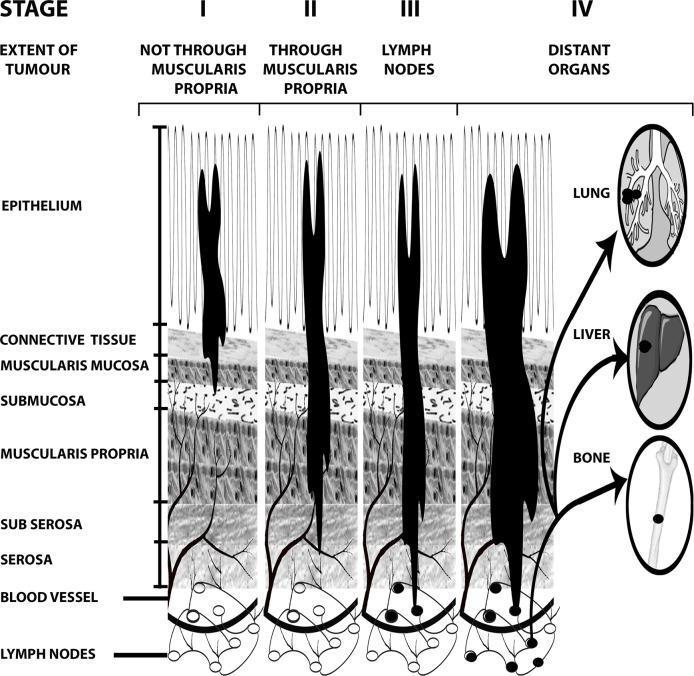
| Stage | Description | Approx. 5-Year Survival |
|---|---|---|
| I | Tumor confined to mucosa/submucosa or muscularis propria (T1–T2, N0, M0) | ~90% |
| II | Extends through muscularis propria into subserosa/serosa; no lymph nodes (T3–T4, N0, M0) | ~70–80% |
| III | Regional lymph node involvement (any T, N1–N2, M0) | ~40–70% |
| IV | Distant metastases (liver, lung, peritoneum) | ~10–15% |
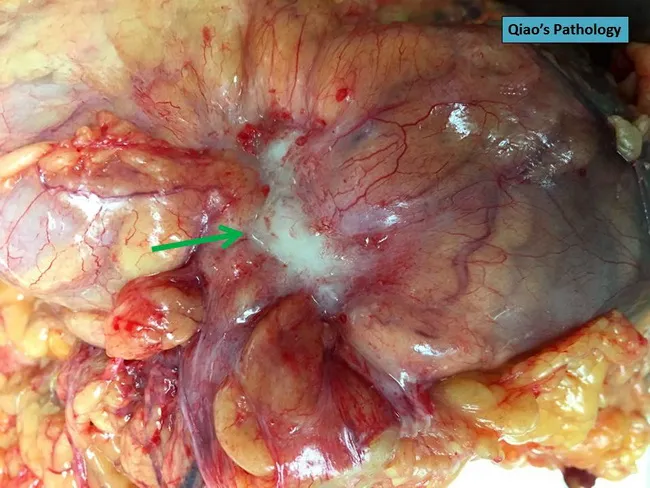
Above: Colorectal adenocarcinoma with serosal puckering indicating T4a invasion (pT4a per TNM staging).
Treatment
Surgery
- Colon cancer: Wide surgical resection with regional lymph node clearance (after bowel preparation)
- Rectal cancer: Abdominoperineal resection (APR) or low anterior resection (LAR); total mesorectal excision (TME) is the standard
Chemotherapy Agents
| Class | Examples |
|---|---|
| Cytotoxic | Irinotecan (topoisomerase I inhibitor), Oxaliplatin |
| Fluoropyrimidines | 5-FU (intravenous), Capecitabine, Tegafur (oral) |
| Anti-EGFR mAbs | Cetuximab (FDA 2004), Panitumumab (FDA 2007) — only for RAS/KRAS wild-type |
| Anti-VEGF/Antiangiogenic | Bevacizumab (anti-VEGF), Ziv-aflibercept, Regorafenib |
| Immunotherapy | Pembrolizumab, Nivolumab — for MSI-High / dMMR tumors |
Key principle: KRAS mutation testing must be performed before anti-EGFR therapy; mutant tumors do not respond and should not receive cetuximab or panitumumab.
Radiation
- Used primarily for rectal cancer — neoadjuvant chemoradiation (before surgery) shrinks the tumor, enables sphincter-sparing surgery, and reduces local recurrence
Screening (USPSTF Recommendations)
| Test | Frequency |
|---|---|
| Annual highly sensitive FOBT or FIT | Every year |
| Multi-target stool DNA (Cologuard) | Every 1–3 years |
| CT colonography | Every 5 years |
| Flexible sigmoidoscopy | Every 5 years (or 10 yr + annual FOBT) |
| Colonoscopy | Every 10 years |
- Average risk: Begin at age 45 (USPSTF 2021 update, lowered from 50)
- Ages 45–75: Routine screening
- Ages 75–85: Selective (individual decision)
- High risk (family history, IBD, genetic syndromes): Earlier and more frequent screening
Prevention
- Aspirin: Long-term use reduces CRC incidence; risks and benefits must be weighed individually
- Diet: Plant-based diet, high fiber, dairy/calcium — all associated with reduced risk
- Lifestyle: Increased physical activity, weight loss, smoking cessation, reduced alcohol
- Colonoscopic polypectomy: Removes adenomas before malignant transformation — the most direct preventive intervention
Key references:
- Henry's Clinical Diagnosis and Management by Laboratory Methods
- Tietz Textbook of Laboratory Medicine, 7th Edition
- Swanson's Family Medicine Review
- Goldman-Cecil Medicine, International Edition
- SEER Cancer Statistics: Colorectal Cancer (2026 estimates)
- WHO Colorectal Cancer Fact Sheet
Astrocytoma
Reading File
Finding Sources
Searching PubMed
"astrocytoma"[MeSH Terms] AND ("classification" OR "treatment")
Searching the Web
astrocytoma WHO 2021 classification IDH grading treatment overview
Reading File
Reading File
Reading File
Searching Images
astrocytoma glioblastoma brain MRI histology pathology
Reading File
Reading File
Reading File
Reading File
I now have comprehensive content from all major textbooks. I'll compose the full response.
Astrocytoma
Astrocytomas are primary brain tumors arising from astrocytes — glial cells that support neurons throughout the CNS. They constitute the largest group of primary glial tumors (gliomas) and encompass a wide biological spectrum, from nearly benign childhood tumors to highly aggressive adult malignancies.
WHO 2021 Classification
The 2021 WHO CNS Tumor Classification (5th Edition) restructured astrocytomas around molecular markers, particularly IDH mutation status, replacing the older purely histological grading system. Key changes:
- The term "anaplastic" is now omitted
- Glioblastoma is now reserved exclusively for IDH-wild-type tumors (Grade 4)
- IDH-mutant astrocytomas are graded CNS WHO Grade 2, 3, or 4 as a continuous entity
- Grade 1 is not used for diffuse astrocytomas (by convention, all diffuse gliomas are considered malignant)
- Molecular features can override histology — e.g., homozygous deletion of CDKN2A/B upgrades a tumor to Grade 4 regardless of histologic appearance
Adams & Victor's Principles of Neurology, 12th Ed.; Robbins & Kumar Basic Pathology
Major Subtypes
1. Pilocytic Astrocytoma (WHO Grade 1)
The most common glioma in children, representing ~20% of all childhood brain tumors. It is circumscribed (non-infiltrating) and considered relatively benign.
Key features:
- Typically arises in the cerebellum, optic pathways, hypothalamus, or brainstem in children; spinal cord involvement possible
- No IDH1/IDH2 mutations
- Molecular driver: KIAA1549-BRAF fusion (80–90% of cerebellar PAs) or BRAF V600E point mutation → constitutive MAPK signaling pathway activation
- NF1 (Neurofibromatosis type 1) is a predisposing condition
- Peak incidence: first two decades of life
Histology: Bipolar cells with long "hairlike" (pilocytic) processes, Rosenthal fibers, eosinophilic granular bodies, microcysts. Necrosis and mitoses are rare.
Imaging: Homogeneously enhancing mass with minimal associated edema; classic appearance is a large cyst with a mural nodule (especially in cerebellum). MRI: T1 hypointense solid component, T2 hyperintense, intense gadolinium enhancement.
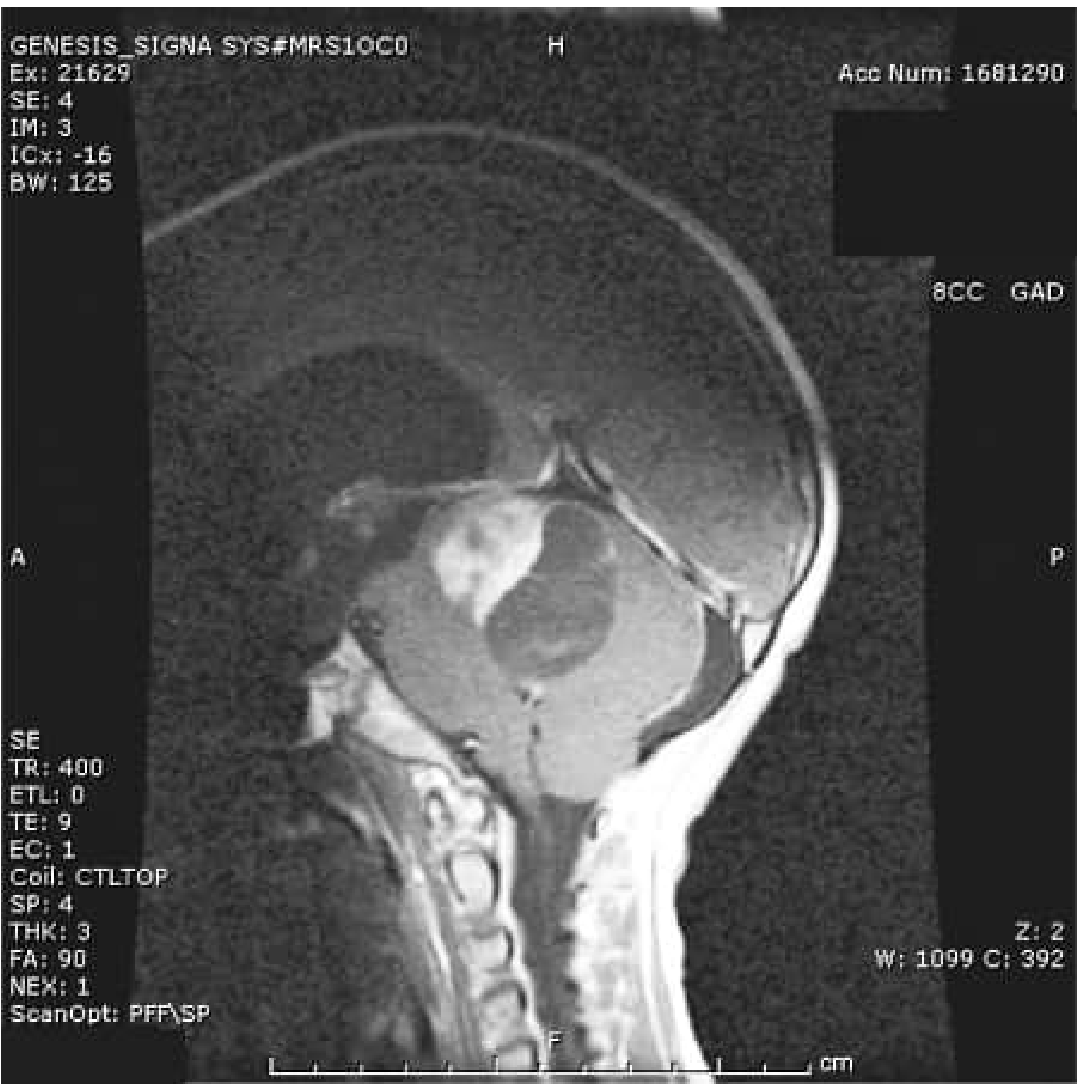
Prognosis: Excellent with complete resection. 5-year survival >90% after surgery. Recurrence can be due to cyst re-expansion rather than solid tumor regrowth.
2. Diffuse Astrocytoma, IDH-Mutant (WHO Grade 2)
- Arises in adults (most common in the 4th–6th decades); also a common pediatric brain tumor (12–18% of all pediatric intracranial tumors)
- Infiltrative — expands through brain parenchyma without a discrete border, tumor cells may infiltrate many centimeters from the main lesion
- Key molecular markers: IDH1 mutation (most common; codon R132H), less often IDH2, plus TP53 and ATRX inactivating mutations
- 80% of adult diffuse astrocytomas harbor IDH1 mutations; IDH mutations are rare in pediatric diffuse astrocytomas (except in late adolescence)
Histology: Mild–moderate increase in glial nuclei, variable nuclear pleomorphism, GFAP-positive fibrillary background. Borders with normal brain are indistinct. No significant mitotic activity at Grade 2.
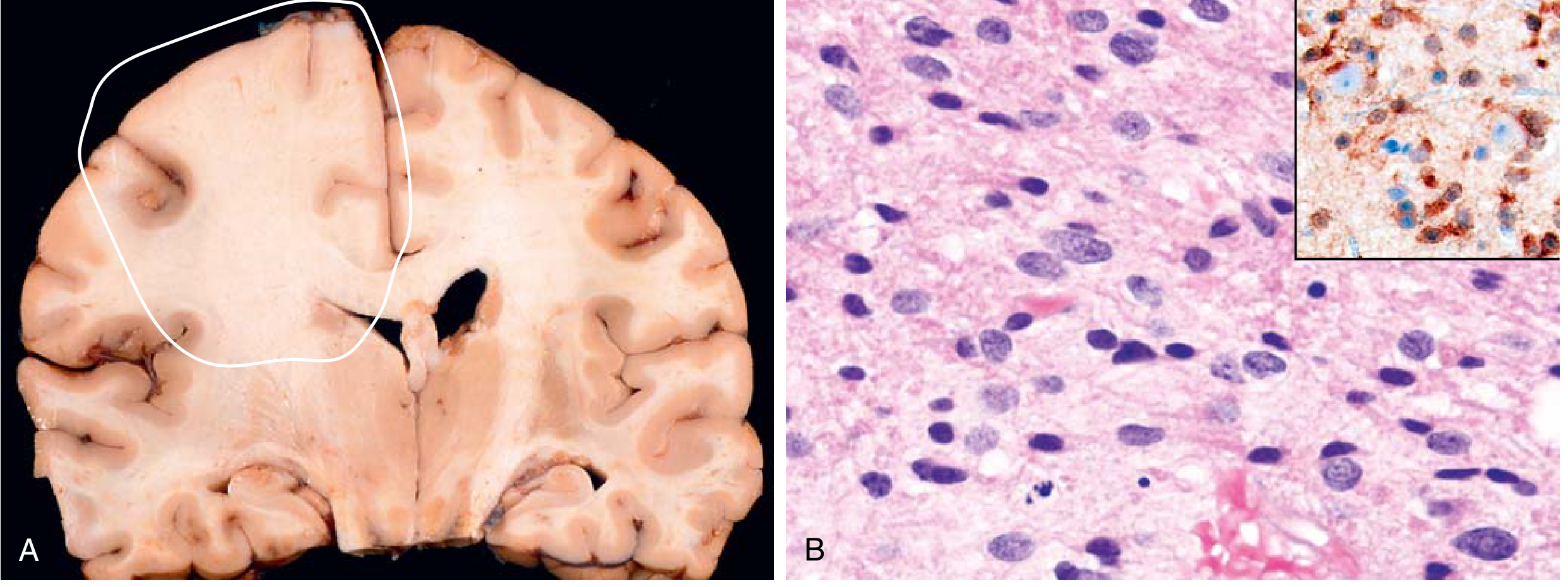
MRI: T1 isointense or hypointense; T2 hyperintense infiltrating mass with poorly defined borders; little or no contrast enhancement. Minimal mass effect.
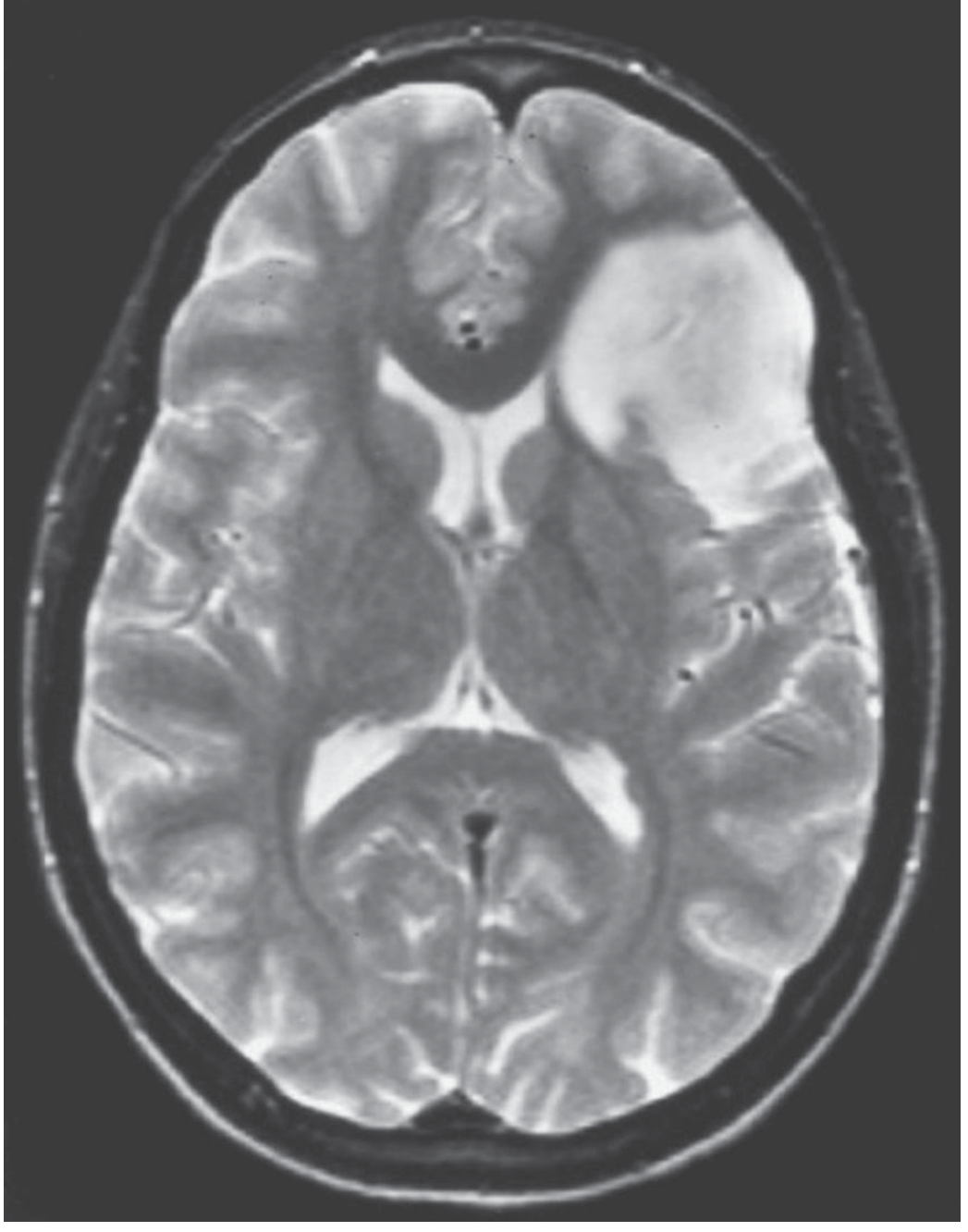
Prognosis: Median overall survival >10 years for Grade 2. Tumors can remain static for years, but ultimately progress.
3. Astrocytoma, IDH-Mutant, WHO Grade 3
- Histologically more cellular than Grade 2, with significant nuclear pleomorphism and mitotic figures
- Same IDH mutation profile as Grade 2 (continuous disease entity)
- More rapid clinical progression
Prognosis: Median survival 5–10 years
4. Astrocytoma, IDH-Mutant, WHO Grade 4
- Distinct from IDH-wild-type glioblastoma in biology, behavior, and prognosis
- Reaches Grade 4 designation by either:
- Histology: microvascular proliferation and/or necrosis
- Molecular: homozygous deletion of CDKN2A and/or CDKN2B (even with lower-grade histology)
- Unlike IDH-wild-type GBM, usually lacks large central necrosis and hemorrhage
- Typically evolves from lower-grade IDH-mutant precursors (secondary glioblastoma pathway)
Prognosis: Median survival ~3 years (significantly better than IDH-wild-type GBM at ~15 months)
5. Glioblastoma, IDH-Wild-Type (WHO Grade 4)
The most aggressive primary brain tumor. Now classified separately from IDH-mutant astrocytomas.
- Arises de novo (primary GBM), predominantly in adults >50 years
- Characterized by marked heterogeneity, rapid growth, extensive infiltration
- Key molecular features: EGFR amplification, PTEN loss, TERT promoter mutation, CDKN2A deletion, chromosome 10 monosomy
- MGMT promoter methylation: predicts benefit from temozolomide chemotherapy; ~45% of GBMs are methylated
- Histology: dense cellularity, marked atypia, brisk mitoses, pseudopalisading necrosis, microvascular proliferation (endothelial hyperplasia)
6. Diffuse Midline Glioma, H3 K27-Altered
A distinct entity in the WHO 2021 classification, predominantly in children:
- Located in the brainstem (DIPG), thalamus, or spinal cord
- Defining mutation: Histone H3 K27M substitution → aberrant DNA methylation, gene expression dysregulation
- Extremely poor prognosis regardless of histological grade; median survival <1 year for DIPG
- G34R/V substitutions in H3 occur in cortical tumors (pediatric)
Bradley & Daroff's Neurology in Clinical Practice
Summary of WHO Grading & Prognosis
| Tumor | WHO Grade | IDH Status | Key Molecular Feature | Median Survival |
|---|---|---|---|---|
| Pilocytic astrocytoma | 1 | Wild-type | BRAF fusion / V600E | >10 yrs (surgical cure possible) |
| Diffuse astrocytoma | 2 | Mutant | IDH1/2, TP53, ATRX | >10 years |
| Astrocytoma | 3 | Mutant | IDH1/2 + mitoses | 5–10 years |
| Astrocytoma | 4 | Mutant | IDH1/2 + CDKN2A/B del. | ~3 years |
| Glioblastoma | 4 | Wild-type | EGFR amp, TERT, PTEN loss | ~15 months |
| Diffuse midline glioma | 4 | Wild-type | H3 K27M | <12 months |
Clinical Presentation
Adults (diffuse/high-grade astrocytomas):
- Seizures — first symptom in ~2/3 of patients with astrocytoma; 60–75% have recurrent seizures
- Focal neurological deficits (location-dependent)
- Headache and signs of raised ICP (relatively late)
- Personality/cognitive change
Children (pilocytic/low-grade):
- Gait unsteadiness, ataxia (cerebellar tumors)
- Nausea and vomiting (obstructive hydrocephalus)
- Visual disturbance (optic pathway tumors)
- Diencephalic syndrome: emaciation with normal linear growth, hyperemesis, hyperkinesis, nystagmus (hypothalamic/optic pathway PAs in infants)
Adams & Victor's Principles of Neurology; Bradley & Daroff's Neurology in Clinical Practice
Imaging
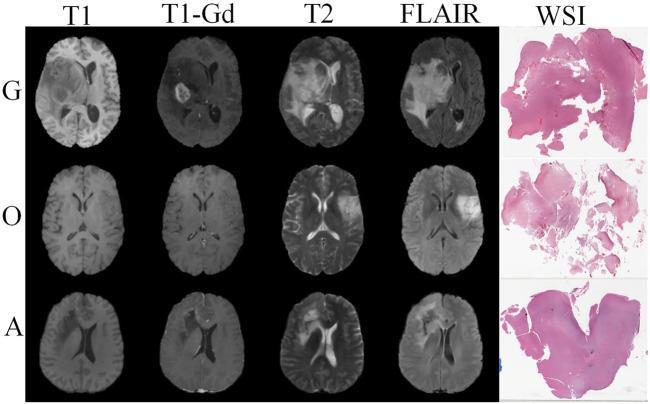
| Feature | Low-grade (Grade 2) | High-grade (Grade 3–4) |
|---|---|---|
| T1 | Iso- to hypointense | Iso- to hypointense |
| T2/FLAIR | Hyperintense | Hyperintense + vasogenic edema |
| Contrast enhancement | Minimal or absent | Irregular ring/nodular enhancement |
| Necrosis | Absent | Present (GBM) |
| Mass effect | Mild | Significant |
| Borders | Ill-defined, infiltrative | Infiltrative ± pseudoprogression |
Spinal astrocytomas (Grainger & Allison):
- Most common intramedullary tumor in children (up to 90%); 30% in adults
- Thoracic cord most common (~70%), often spanning multiple levels
- T1 iso/hypointense, T2 hyperintense; low-grade tumors often don't enhance; high-grade show patchy enhancement
- Cystic degeneration in 60%; associated syringomyelia possible
Treatment
Surgery
- Maximal safe resection is the cornerstone for all grades
- Low-grade astrocytomas: early surgery improves survival vs. watchful waiting (Norwegian series data)
- Pilocytic astrocytomas: surgical cure is achievable with complete resection
- Modern brain mapping (language, motor) enables safer resection of eloquent area tumors
- For spinal astrocytomas: biopsy only is typical (infiltrative; complete resection not usually possible)
Radiotherapy
- Standard for Grade 3–4 tumors post-surgery
- For GBM: 60 Gy in 30 fractions (Stupp protocol)
- For low-grade (Grade 2) in high-risk patients (incomplete resection, older age): adjuvant RT improves PFS
Chemotherapy
- Temozolomide (TMZ): alkylating agent; standard of care for GBM (Stupp protocol: concurrent + adjuvant TMZ with RT)
- MGMT promoter methylation predicts TMZ benefit — methylated tumors respond significantly better
- PCV regimen (procarbazine, CCNU/lomustine, vincristine): used in lower-grade IDH-mutant gliomas (Grade 2–3)
- Bevacizumab (anti-VEGF): used in recurrent GBM
Targeted / Emerging Therapies
- BRAF inhibitors (dabrafenib + trametinib): active in BRAF V600E–mutant astrocytomas
- MEK inhibitors: being trialed for BRAF-fused (non-V600E) pediatric low-grade gliomas
- IDH inhibitors (ivosidenib, enasidenib): under investigation for IDH-mutant gliomas
- CAR-T cells: active investigation for GBM (targeting EGFRvIII, IL13Rα2, GD2) — multiple systematic reviews 2024–2025 (PMID 39000281)
Tumor Treating Fields (TTFields)
- Alternating electric field device (Optune®): added to temozolomide for newly diagnosed GBM; modestly extends survival
Hereditary Predisposition Syndromes
| Syndrome | Gene | Associated Tumor |
|---|---|---|
| Neurofibromatosis type 1 (NF1) | NF1 | Pilocytic astrocytoma (optic pathway) |
| Li-Fraumeni syndrome (LFS) | TP53 | High-grade astrocytomas |
| Constitutional MMR Deficiency (cMMRD) | MLH1, MSH2, MSH6, PMS2 | Pediatric HGAs; GI/hematologic cancers |
| Turcot syndrome (Type 1) | MMR genes | GBM + colorectal polyposis |
Key references:
- Adams & Victor's Principles of Neurology, 12th Edition
- Robbins & Kumar Basic Pathology
- Bradley & Daroff's Neurology in Clinical Practice
- Grainger & Allison's Diagnostic Radiology
- WHO CNS Tumor Classification 2021 (PMC overview)
- CAR-T in GBM systematic review — PMID 39000281 (Int J Mol Sci, 2024)
This is a shared conversation. Sign in to Orris to start your own chat.