Punctate keratoderna
Reading File
Finding Sources
Finding Sources
Searching PubMed
punctate keratoderma
Reading File
Reading File
Reading File
Loading Image
Reading File
Reading File
I now have comprehensive information. Let me compile the full answer.
Punctate Keratoderma (Punctate Palmoplantar Keratoderma, PPKP)
Punctate palmoplantar keratoderma (punctate PPK) is a group of rare hereditary (and occasionally acquired) disorders characterized by discrete, small keratotic papules or spines confined to the palms and soles, in contrast to diffuse or striate forms. Three main types are recognized.
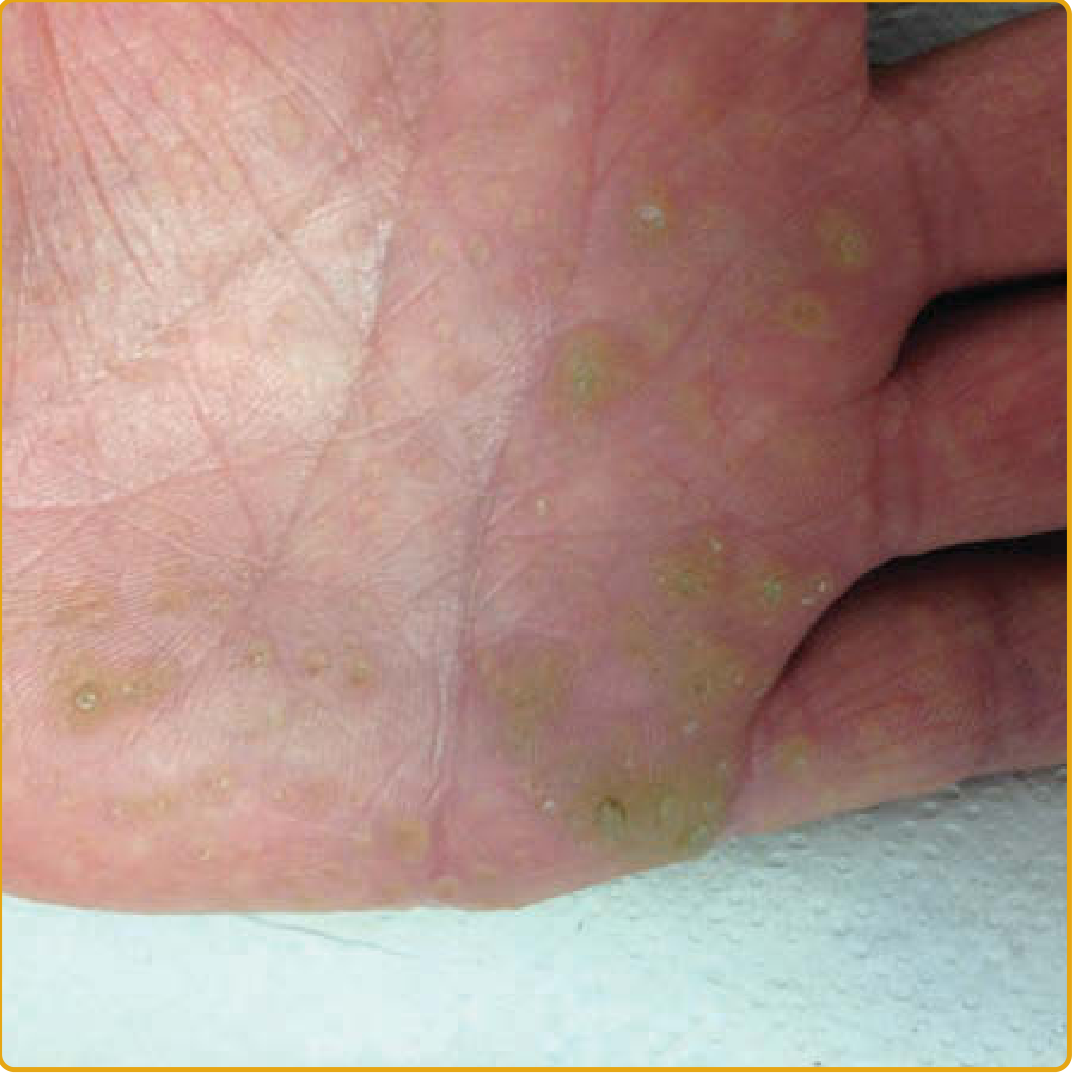
Punctate PPK type I (AAGAB mutation): multiple hyperkeratotic, centrally indented, yellow-to-brown papules irregularly distributed over the palmar skin. (Fitzpatrick's Dermatology, Vol. 1 & 2)
Classification
Type I - Buschke-Fischer-Brauer (BFB) Type
(OMIM #148600 [PPKP1A] / #614936 [PPKP1B])
- Synonyms: Keratosis punctata palmoplantaris, Keratoma dissipatum, Disseminated clavus
- Inheritance: Autosomal dominant
- Incidence: ~1-3 per 100,000
Genetics and pathogenesis:
- AAGAB mutations (PPKP1A) - encodes the alpha- and gamma-adaptin-binding protein p34, involved in intracellular trafficking of clathrin-coated vesicles. The defect impairs endocytic recycling of receptor tyrosine kinases (EGFR, Axl), leading to increased signaling and cellular proliferation. On electron microscopy: large number of small vesicles + dilated Golgi apparatus.
- COL14A1 mutations (PPKP1B) - a missense mutation in collagen type XIV alpha-1 chain gene was identified in a large Chinese family (incomplete penetrance).
Clinical features:
- Onset: first or second decade (but can range from childhood to sixth decade)
- Multiple hyperkeratotic, centrally indented, yellow-to-brown papules distributed irregularly over palmoplantar skin
- The central keratotic core is initially translucent, may become opaque or verrucous over time; a pit/depression remains after removal
- Lesions increase in size and number with age; can coalesce into plaques over pressure areas
- May be painful or tender
- Worsened by mechanical stress
- Wood lamp: white fluorescence
- Dermoscopy: well-demarcated, structureless yellow-orange areas surrounded by whitish halo; no dotted vessels (distinguishes from verruca)
- Genetic anticipation (earlier onset in successive generations) has been reported
Histopathology:
- Slight epidermal depression with overlying column of compact orthohyperkeratosis
- Focal hypogranulosis with parakeratosis; elongated, curved rete ridges
- No cytologic features of HPV infection
Special mention - Punctate keratoses of palmar creases:
A variant seen frequently in adults of African ancestry - keratotic papules along palmar creases (digits and soles also affected), worsened by mechanical stress. The relationship to classic punctate PPK is not fully established.
Type II - Spiny Keratoderma (Porokeratotic Type)
(OMIM #175860)
- Synonyms: Palmoplantar keratoderma punctata type 2, Music box spine keratoderma, Filiform hyperkeratosis, Digitate keratosis
- Inheritance: Autosomal dominant (molecular etiology unknown)
- Onset: puberty or early 20s
Clinical features:
- Multiple, tiny, firmly attached, skin-colored to yellow, asymptomatic keratotic spines projecting from palms and soles
- Discomfort caused by tendency to catch on clothing
- May extend onto dorsal and lateral surfaces of fingers
- Facial sebaceous hypoplasia reported in males
- Wood lamp: white fluorescence resembling "stars under moonlight"
Acquired forms of spiny PPK (usually onset >50 years) are associated with:
- Internal malignancies (visceral carcinoma, melanoma, leukemia, multiple myeloma)
- Hyperlipidemia type IV
- Chronic kidney disease, polycystic kidney disease with liver cysts
Histopathology:
- Epidermal depression with overlying column of parakeratosis (in contrast to Type I's orthohyperkeratosis)
- Stratum granulosum is preserved
- No vacuolization, pleomorphism, or dyskeratosis of keratinocytes (distinguishes from porokeratosis)
Type III - Acrokeratoelastoidosis (AKE) of Costa
(OMIM #101850)
- Inheritance: Autosomal dominant; rare, no racial predilection
- Onset: childhood or adolescence (infancy or adulthood also reported)
Clinical features:
- Asymptomatic, round-to-oval, whitish-to-yellow, translucent papules; rarely nodules and plaques with hyperkeratotic surface or umbilication
- Predilection for thenar, hypothenar areas, pressure sites on palms and soles
- Linear pattern or "paving stones" arrangement along radial/ulnar margins of hands
- Severe cases: dorsal hands and feet, wrists, ankles (including knuckle pads)
- Aquagenic PPK, nail dystrophy, and hyperhidrosis may be associated
Differential Diagnosis
| Condition | Key distinguishing feature |
|---|---|
| Verruca vulgaris | Multiple bleeding points on paring; dotted vessels on dermoscopy; HPV features on histology |
| Punctate porokeratosis | Cornoid lamella on histology (instead of orthohyperkeratosis) |
| Arsenic/dioxin keratoses | Environmental exposure history |
| Darier disease | Histology: corps ronds and grains |
| Acrokeratosis verruciformis of Hopf | Dorsal hands, more verrucous |
| Cowden syndrome | Trichilemmomas, mucosal lesions, systemic findings |
| Pitted keratolysis | Bacterial, malodour, pits not true keratoses |
Malignancy Association
A 2023 systematic review (PMID 37705065) specifically examined whether PPKP type I is associated with malignancy. Older reports suggested possible links with breast and colon carcinomas, but it remains unclear whether there is truly an excess of malignancies in affected individuals, as noted in [Dermatology 2-Volume Set 5e, p. 1128].
Treatment
- Comfortable footwear and mechanical paring/debridement for symptomatic relief
- Topical retinoids and keratolytic ointments have limited efficacy
- Oral retinoids have been tried
- No curative treatment; management is largely symptomatic
Sources: Dermatology 2-Volume Set 5e, pp. 1127-1128 | Fitzpatrick's Dermatology, Vol. 1 & 2, pp. 873-874 | Andrews' Diseases of the Skin Clinical Dermatology, p. 250
This is a shared conversation. Sign in to Orris to start your own chat.